

Определение генетического полиморфизма расшифровка: Генетический риск развития тромбофилии (расширенный)
Генетический риск развития тромбофилии (расширенный)
Комплексный генетический анализ, который позволяет определить риск тромбофилии. Он представляет собой молекулярно-генетическое исследование генов факторов свертываемости крови, тромбоцитарных рецепторов, фибринолиза, обмена фолиевой кислоты, изменение активности которых напрямую или опосредованно обуславливает склонность к повышенному тромбообразованию.
Какой биоматериал можно использовать для исследования?
Буккальный (щечный) эпителий, венозную кровь.
Как правильно подготовиться к исследованию?
Подготовки не требуется.
Подробнее об исследовании
В результате различных патологических процессов в сосудах могут образоваться тромбы, которые блокируют кровоток. Это самое частое и неблагоприятное проявление наследственной тромбофилии – повышенной склонности к тромбообразованию, связанной с определенными генетическими дефектами. Она может приводить к развитию артериальных и венозных тромбозов, которые в свою очередь зачастую являются причиной инфаркта миокарда, ишемической болезни сердца, инсульта, тромбоэмболии легочной артерии и др.
В систему гемостаза входят факторы свертывающей и противосвертывающей систем крови. В нормальном состоянии они находятся в равновесии и обеспечивают физиологические свойства крови, не допуская повышенного тромбообразования или, наоборот, кровоточивости. Но при воздействии внешних или внутренних факторов это равновесие может нарушаться.
В развитии наследственной тромбофилии, как правило, принимают участие гены факторов свертывания крови и фибринолиза, а также гены ферментов, контролирующих обмен фолиевой кислоты. Нарушения в этом обмене могут привести к тромботическим и атеросклеротическим поражениям сосудов (через повышение уровня гомоцистеина в крови).
Наиболее значимым нарушением, ведущим к тромбофилии, является мутация в гене фактора свертываемости 5 (F5), ее еще называют Лейденской. Она проявляется устойчивостью фактора 5 к активированному протеину С и увеличением скорости образования тромбина, в результате чего и происходит усиление процессов свертываемости крови. Также важную роль в развитии тромбофилии играет мутация в гене протромбина (F2), связанная с повышением уровня синтеза данного фактора свертываемости. При наличии этих мутаций риск тромбозов значительно возрастает, особенно за счет провоцирующих факторов: приема оральных контрацептивов, избыточного веса, гиподинамии и т. д.
Также важную роль в развитии тромбофилии играет мутация в гене протромбина (F2), связанная с повышением уровня синтеза данного фактора свертываемости. При наличии этих мутаций риск тромбозов значительно возрастает, особенно за счет провоцирующих факторов: приема оральных контрацептивов, избыточного веса, гиподинамии и т. д.
У носительниц таких мутаций высока вероятность неблагоприятного течения беременности, например невынашивания беременности, задержки внутриутробного развития плода.
Предрасположенность к тромбозам может быть также обусловлена мутацией гена FGB, кодирующего бета-субъединицу фибриногена (генетический маркер FGB (-455GA). Результатом является повышение синтеза фибриногена, вследствие чего возрастает риск периферического и коронарного тромбоза, риск тромбоэмболических осложнений во время беременности, при родах и в послеродовом периоде.
Среди факторов, повышающих риск развития тромбоза, очень важны гены тромбоцитарных рецепторов. В данном исследовании проводится анализ генетического маркера гена тромбоцитарного рецептора к коллагену (ITGA2 807 C>T) и фибриногену (ITGB3 1565T>C). При дефекте гена рецептора к коллагену усиливается прилипание тромбоцитов к эндотелию сосудов и к друг к другу, что ведет к повышенному тромбообразованию. При анализе генетического маркера ITGB3 1565T>C возможно выявить эффективность или неэффективность антиагрегантной терапии аспирином. При нарушениях, обусловленных мутациями в этих генах, повышается риск тромбозов, инфаркта миокарда, ишемического инсульта.
В данном исследовании проводится анализ генетического маркера гена тромбоцитарного рецептора к коллагену (ITGA2 807 C>T) и фибриногену (ITGB3 1565T>C). При дефекте гена рецептора к коллагену усиливается прилипание тромбоцитов к эндотелию сосудов и к друг к другу, что ведет к повышенному тромбообразованию. При анализе генетического маркера ITGB3 1565T>C возможно выявить эффективность или неэффективность антиагрегантной терапии аспирином. При нарушениях, обусловленных мутациями в этих генах, повышается риск тромбозов, инфаркта миокарда, ишемического инсульта.
С тромбофилией могут быть связаны не только нарушения свертывающей системы крови, но и мутации генов фибринолитической системы. Генетический маркер SERPINE1 (-675 5G>4G) – ингибитор активатора плазминогена – основного компонента антисвертывающей системы крови. Неблагоприятный вариант этого маркера приводит к ослаблению фибринолитической активности крови и, как следствие, повышает риск сосудистых осложнений, различных тромбоэмболий. Мутация гена SERPINE1 также отмечается при некоторых осложнениях беременности (невынашивание беременности, задержка развития плода).
Мутация гена SERPINE1 также отмечается при некоторых осложнениях беременности (невынашивание беременности, задержка развития плода).
Кроме мутаций факторов свертывающей и противосвертывающей систем, как значимую причину тромбофилии рассматривают повышенный уровень гомоцистеина. При чрезмерном накоплении он оказывает токсическое действие на эндотелий сосудов, поражает сосудистую стенку. В месте повреждения образуются тромбы, там же может осесть избыточный холестерин. Эти процессы приводят к закупориванию сосудов. Избыточное содержание гомоцистеина (гипергомоцистеинемия) увеличивает вероятность развития тромбозов в кровеносных сосудах (как в артериях, так и в венах). Одной из причин повышения уровня гомоцистеина является снижение активности ферментов, обеспечивающих его обмен (в исследование включен ген MTHFR). Помимо генетического риска развития гипергомоцистеинемии и ассоциированных с ней заболеваний, наличие изменений в данном гене позволяет определить предрасположенность и к неблагоприятному течению беременности (фетоплацентарной недостаточности, незаращению нервной трубки и другим осложнениям для плода). При изменениях в фолатном цикле в качестве профилактики назначаются фолиевая кислота и витамины В6, В12. Продолжительность терапии и дозировка препаратов может определяться на основании генотипа, уровня гомоцистеина и особенностей сопутствующих факторов риска у пациента.
При изменениях в фолатном цикле в качестве профилактики назначаются фолиевая кислота и витамины В6, В12. Продолжительность терапии и дозировка препаратов может определяться на основании генотипа, уровня гомоцистеина и особенностей сопутствующих факторов риска у пациента.
Заподозрить наследственную предрасположенность к тромбофилии возможно при семейной и/или личной истории тромботических заболеваний (тромбоз глубоких вен, варикозная болезнь и др.) и также в акушерской практике – при тромбоэмболических осложнениях у женщин во время беременности, в послеродовом периоде.
Комплексное молекулярно-генетическое исследование позволяет оценить генетический риск тробофилии. Зная о генетической предрасположенности можно предотвратить своевременными профилактическими мероприятиями развитие сердечно-сосудистых нарушений..
Факторы риска развития тромбофилии:
- постельный режим (более 3 суток), длительная иммобилизация, долгие статические нагрузки, в том числе связанные с работой, малоподвижный образ жизни;
- применение пероральных контрацептивов, содержащих эстрогены;
- избыточная масса тела;
- венозные тромбоэмболические осложнения в анамнезе;
- катетер в центральной вене;
- обезвоживание;
- хирургические вмешательства;
- травма;
- курение;
- онкологические заболевания;
- беременность;
- сопутствующие сердечно-сосудистые заболевания, злокачественные новообразования.

Когда назначается исследование?
- При наличии тромбоэмболии в семейном анамнезе.
- При наличии тромбоза в анамнезе.
- При тромбозе в возрасте до 50 лет, повторных тромбозах.
- В случае тромбоза в любом возрасте в сочетании с отягощенным семейным анамнезом по тромбоэмболии (тромбоэмболия лёгочной артерии), в том числе при тромбозах других локализаций (сосудов мозга, портальных вен).
- При тромбозе без очевидных факторов риска в возрасте старше 50 лет.
- В случае применения гормональных контрацептивов или заместительной гормональной терапии у женщин: 1) имеющих тромбозы в анамнезе, 2) у родственников 1-й степени родства которых были тромбозы или наследственная тромбофилия.
- При осложненном акушерском анамнезе (невынашивании беременности, фетоплацентарной недостаточности, тромбозах во время беременности и в раннем послеродовом периоде и др.).
- При планировании беременности женщинами, страдающими тромбозами (или в случае тромбоза у их родственников 1-й степени родства).
- При таких условиях высокого риска, как полостные оперативные вмешательства, длительная иммобилизация, постоянные статические нагрузки, малоподвижный образ жизни.
- При сердечно-сосудистых заболеваниях в семейном анамнезе (случаи ранних инфарктов и инсультов).
- При оценке риска тромботических осложнений у больных со злокачественными новообразованиями.
Что означают результаты?
По результатам комплексного исследования 10 значимых генетических маркеров выдается заключение врача-генетика, которое позволит оценить риск тромбофилии, спрогнозировать развитие таких заболеваний как тромбоз, тромбоэмболия, инфаркт, или вероятность осложнений, связанных с нарушением гемостаза, при беременности, выбрать направления оптимальной профилактики, а при уже имеющихся клинических проявлениях детально разобраться в их причинах.
|
Определение полиморфизма в геноме человека (формат PCR—RT)
|
|
|
Определение генетического полиморфизма предрасположенности к артериальной гипертензии
|
|
|
|
Комплексное генетическое исследование, которое позволяет спрогнозировать риск гипертонии и связанных с ней заболеваний, выявить патологию на ранних сроках.
|
|
Определение генетического полиморфизма предрасположенности к тромбообразованию
|
|
|
|
Комплексный генетический анализ, который позволяет определить риск тромбофилии. Он представляет собой молекулярно-генетическое исследование генов факторов свертываемости крови, тромбоцитарных рецепторов, фибринолиза, обмена фолиевой кислоты, изменение активности которых напрямую или опосредованно обуславливает склонность к повышенному тромбообразованию.
|
|
Определение генетического полиморфизма предрасположенности к нарушениям метаболизма фолатного цикла
|
|
|
|
Комплексное генетическое исследование, которое позволяет определить риск сердечно-сосудистых заболеваний, а также патологии развития плода, связанных с нарушением обмена фолиевой кислоты и гипергомоцистеинемией.
|
|
Определение фармакогенетических маркеров метаболизма лекарственных препаратов
|
|
|
|
Фармакогенетическое исследование, направленное на определение индивидуальной безопасной дозы препарата варфарина (антикоагулянта непрямого действия). Проводится анализ на наличие мутаций по трем генетическим маркерам, связанным с повышенной чувствительностью к варфарину. Выявление оптимальной дозы варфарина позволяет сократить срок подбора его дозы, а также снизить риск геморрагических осложнений.
Генетическое исследование, которое позволяет определить эффективность применения препарата «Клопидогрел» на основании активности фермента CYP2C19.
|
|
|
|
|
Определение генетического полиморфизма предрасположенности к раку молочной железы (BRCA ½)
|
|
|
|
Генетическое комплексное исследование, которое позволяет определить наследственную предрасположенность к раку молочной железы и яичников.
|
|
Определение генетического полиморфизма предрасположенности к первичной лактазной недостаточности
|
|
|
|
Лактозная непереносимость связана с генетически обусловленным снижением активности лактазы (фермента, необходимого для усвоения лактозы) и проявляется неспецифическими симптомами, развивающимися после потребления продуктов, содержащих молочный сахар. Возникает после периода грудного вскармливания.
|
|
Определение генетического полиморфизма интерлейкина ILВ28
|
|
|
|
Это прогностическое тестирование, дающее возможность оценить успешность (вероятность успешности) курса лечения хронического гепатита С.
|
|
Определение генетического полиморфизма компонентов главного комплекса гистосовместимости человека HLA
|
|
|
|
Выявление гена гистосовместимости HLA-B27. Определение предрасположенности к развитию спондилоартропатий (в т.ч. анкилозирующего спондилита — болезнь Бехтерева)
|
|
|
Комплексное исследование, определяющее генетическую гистосовместимость партнеров. Различие супругов по вариантам генов HLA считается одним из важных условий успешного наступления и вынашивания беременности. Сходство супругов между собой по вариантам генов HLA ведет к повышению вероятности появления плода с двойным набором одинаковых вариантов генов, то есть HLA-гомозигот, или полному совпадению плода и матери по HLA генотипу, что является неблагоприятным фактором, следствием чего могут стать репродуктивные потери.
|
|
Определение генетических факторов мужского бесплодия
|
|
|
|
Комплексное исследование, которое позволяет определить ведущие генетические причины мужского бесплодия и выбрать соответствующую тактику лечения пациента.
В исследование включено выявление делеций в области локуса AZF, влияющих на сперматогенез, и определение количества CAG-повторов в гене AR, связанных с изменением чувствительности к андрогенам.
|
|
Определение генетики плода
|
|
|
|
Определение гена резус-фактора плода в крови матери
|
 Также оно помогает разобраться в причинах уже имеющейся патологии, выбрать направления оптимальной профилактики и персональной медикаментозной терапии. В анализ включены основные генетические маркеры, участвующие в регуляции артериального давления.
Также оно помогает разобраться в причинах уже имеющейся патологии, выбрать направления оптимальной профилактики и персональной медикаментозной терапии. В анализ включены основные генетические маркеры, участвующие в регуляции артериального давления. Анализ включает в себя молекулярно-генетическое исследование генов ферментов, изменение активности которых приводит к повышению уровня гомоцистеина в крови.
Анализ включает в себя молекулярно-генетическое исследование генов ферментов, изменение активности которых приводит к повышению уровня гомоцистеина в крови. Оно представляет собой анализ наиболее распространенных генетических маркеров (85 % среди всех наследственных форм рака молочной железы и яичников), наличие которых указывает на повышенный риск развития онкологического заболевания.
Оно представляет собой анализ наиболее распространенных генетических маркеров (85 % среди всех наследственных форм рака молочной железы и яичников), наличие которых указывает на повышенный риск развития онкологического заболевания. Исследованию подлежат генетические полиморфизмы гена IL28B расположенные в локусах rs12979860/ rs8099917.
Исследованию подлежат генетические полиморфизмы гена IL28B расположенные в локусах rs12979860/ rs8099917. В связи с этим, HLA-типирование используют для диагностики причин невынашивания беременности и бесплодия. При репродуктивных нарушениях важно количество совпадений вариантов генов HLA II класса у супругов: чем их меньше, тем выше вероятность наступления беременности. Генотипирование HLA класса II включает в себя определение конкретных вариантов генов из всех возможных. В исследование включено 3 локуса гена HLA: HLA-DRB1, HLA-DQA1 и HLA-DQB1.
В связи с этим, HLA-типирование используют для диагностики причин невынашивания беременности и бесплодия. При репродуктивных нарушениях важно количество совпадений вариантов генов HLA II класса у супругов: чем их меньше, тем выше вероятность наступления беременности. Генотипирование HLA класса II включает в себя определение конкретных вариантов генов из всех возможных. В исследование включено 3 локуса гена HLA: HLA-DRB1, HLA-DQA1 и HLA-DQB1.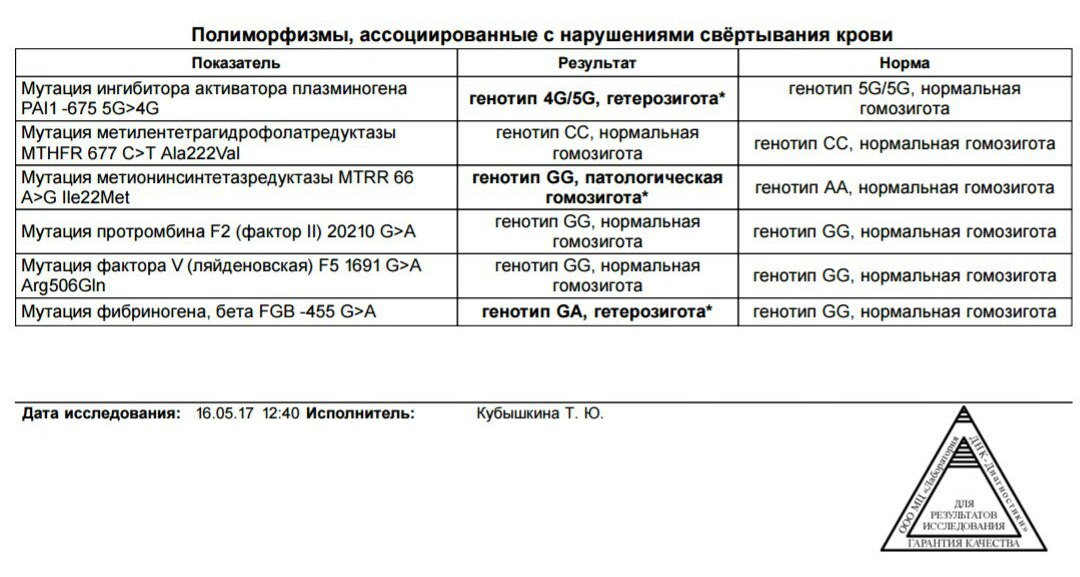
Анализы в KDL. Генетический риск нарушений системы свертывания
Данное исследование представляет собой выявление полиморфизмов в генах, связанных с наследственной тромбофилией и другими нарушениями системы свертывания крови. Тромбофилия – это повышенная склонность к случайному (без видимых причин) тромбозу глубоких вен и тромбоэмболическим осложнениям. Тромбоз может возникнуть при нарушениях кровообращения (застой крови), повышенной способности крови образовывать тромбы (гиперкоагуляции), повреждениях сосудистой стенки и при сочетании этих факторов. Гиперкоагуляция может быть обусловлена наличием генетической предрасположенности к тромбофилии (наиболее часто встречаются полиморфизмы в генах F2 и F5), которую позволяет выявить этот анализ.
В каких случаях обычно назначают исследование?
Анализ рекомендован следующим категориям пациентов:
- пациенты с семейной историей тромбофилии и других венозных осложнений
- пациенты с наличием в семейной истории сердечно- сосудистых событий (инфарктов и инсультов) у близких родственников в возрасте до 50 лет
- женщины, планирующие прием комбинированных
- пациенты с тромбозами любой локализации в возрасте до 50 лет, даже при наличии предрасполагающих факторов
- пациенты с тромбозами необычных мест (тромбоз мозговых синусов, мезентериальные, печеночные вены)
- пациенты с хроническими заболеваниями вен, в частности с тромбофлебитами
- женщины, планирующие прием комбинированных оральных контрацептивов
- женщины, планирующие гормональную заместительную терапию
- женщины с привычным невынашиванием беременности.
Что именно определяется в процессе анализа?
В данном исследовании выявляются полиморфизмы следующих генов:
- F2 — ген протромбина.

- F5 – ген фактора V свертывания крови.
- F7 – ген проконвертина.
- F13A1 – ген, кодирующий A-субъединицу XIII фактора свертывания.
- FGB – ген, кодирующий бета- субъединицу фибриногена.
- ITGA2 – ген, кодирующий тромбоцитарный гликопротеин Iа.
- ITGB3 – ген, кодирующий белок интегрин бета- 3, являющийся компонентом тромбоцитарного гликопротеина IIb/IIIа.
- SERPINE1 (PAI-I)– ген ингибитора активатора плазминогена 1 типа.
Что означают результаты теста?
В качестве заключения выдается генетическая карта здоровья, составленная профессиональным врачом-генетиком нашей лаборатории. В ней представлены результаты анализа с расшифровкой, дано подробное описание исследуемых полиморфизмов и их влияния на риски развития нарушений системы свертывания крови, сердечно-сосудистых заболеваний и патологий беременности, советы по проведению дополнительных исследований и подробные клинические рекомендации для лечащего врача.
Результаты анализа интерпретируются лечащим врачом. Опираясь на них, врач может скорректировать питание и образ жизни пациента, назначить лекарственные препараты и дополнительные исследования.
Сроки выполнения теста.
Результат генетического исследования можно получить спустя 10-11 дней после взятия крови.
Как подготовиться к анализу?
Следует придерживаться общих правил подготовки к взятию крови из вены. Кровь можно сдавать не ранее, чем через 3 часа после приема пищи в течение дня, или утром натощак. Чистую воду можно пить в обычном режиме.
Наследственная тромбофилия
Склонность к патологическому свертыванию крови, или тромбофилия может обнаруживаться у пациентов, столкнувшихся с опасным состоянием — тромбозом 1. И у многих из них склонность к повышенному тромбообразованию передалась по наследству. Почему развивается наследственная тромбофилия. Какие симптомы появляются при данном состоянии, как ее диагностировать и лечить?
Генетические факторы
Тромбофилия, которая передается по наследству, — генетически обусловленная склонность к формированию тромбов. При сочетании двух и более нарушений в системе свертывания выраженные тромботические расстройства могут развиваться уже в раннем детстве. Однако чаще наследственное повышение свертываемости крови вызвано одним нарушением, которое выявляется случайно при лабораторном исследовании крови 2.
При сочетании двух и более нарушений в системе свертывания выраженные тромботические расстройства могут развиваться уже в раннем детстве. Однако чаще наследственное повышение свертываемости крови вызвано одним нарушением, которое выявляется случайно при лабораторном исследовании крови 2.
Выделяют два основных типа тромбофилий: гематогенные, связанные с изменениями свертывающей системы крови, и негематогенные. На сегодня известны как минимум 20 генетических факторов, связанных с развитием заболевания. Их разделяют на три группы2:
-
Установленные:
- фактор V Лейден — мутация, при которой из-за замены аминокислот в белковой цепи V фактора свертывания он становится устойчивым к действию одного из основных противосвертывающих агентов, активированного протеина С.
- Мутация G20210А протромбина II, при которой изменяется уровень фактора свертывания крови II протромбина.
- Дефицит протеина С, инактивирующего факторы свертывания крови Va и VIIIa.
 Встречается реже, чем две предыдущие мутации, обычно приводит к развитию семейного венозного тромбоза.
Встречается реже, чем две предыдущие мутации, обычно приводит к развитию семейного венозного тромбоза. - Дефицит протеина S — кофактора протеина С, который усиливает его противосвертывающую и фибринолитическую активность.
- • Дефицит антитромбина, основного белкового фактора, который угнетает свертывание крови. Передается по аутосомно-доминантному типу, то есть проявляется только при наличии хотя бы одного дефектного гена.
-
Неопределённые:
- Дисфибриногенемия — состояние, при котором содержание фибриногена соответствует норме, однако сама молекула фибриногена изменена. Чаще передается по аутосомному типу наследования, то есть для проявления заболевания дефектный ген должен передаться от обоих родителей. Обычно проявляется умеренными кровотечениями на фоне травмы или хирургического вмешательства.
- Гипергомоцистеинемия. Может быть одновременно и наследственным, и приобретенным нарушением 2.
 Связана с редкими генетическими поломками, которые приводят к повышению концентрации гомоцистеина в моче, плазме крови. Гомоцистеин проявляет выраженный токсический эффект, повреждая внутреннюю поверхность сосудов, значительно увеличивая риск тромбозов.
Связана с редкими генетическими поломками, которые приводят к повышению концентрации гомоцистеина в моче, плазме крови. Гомоцистеин проявляет выраженный токсический эффект, повреждая внутреннюю поверхность сосудов, значительно увеличивая риск тромбозов.
-
Редкие.
К редким факторам тромбофилии относятся повышение концентрации факторов свертывания крови VIII, IX, XI, VII, XII, дефицит плазминогена, активация тканевого плазминогена, повышение липопротеина А, полиморфизм гликопротеина тромбоцитов, дефект гена тромбомодулина, другие факторы.
Как проявляется тромбофилия?
Наличие тромбофилии можно заподозрить при следующих состояниях 2:
- Повторная венозная тромбоэмболия
- Венозный тромбоз в возрасте младше 40 лет
- Венозная тромбоэмболия у родственников
- Тромбоз необычной локализации, например, брыжеечной вены, почечной вены, печени, церебральный тромбоз.
Необходимо отметить, что чаще всего тромбофилия приводит к тромбозу вен, однако недостаточность протеинов С, S и антитромбина могут проявляться также тромбозами в артериях. Последние становятся причиной инфаркта миокарда и острого нарушения мозгового кровообращения — инсульта.
Последние становятся причиной инфаркта миокарда и острого нарушения мозгового кровообращения — инсульта.
Диагностика тромбофилии
Своевременная диагностика генетической (наследственной) тромбофилии играет важную роль в прогнозе заболевания. Раннее выявление заболевания позволяет внести необходимые коррективы в образ жизни и таким образом предупредить патологическое тромбообразование.
Поскольку передающаяся по наследству тромбофилия может быть связана с различными генетическими факторами, диагностика ее носит комплексный характер. Задача врача — подтвердить или опровергнуть наличие мутаций, связанных с нарушением свертывающей активности крови.
К числу анализов, которые придется сдать при подозрениях на наследственную тромбофилию, относятся3:
Развернутая коагулограмма с определением уровня факторов свертывания крови, физиологических антикоагулянтов.
Коагулограмма позволяет выявить отклонения в системе гемостаза и определить тип тромбофилии, недостаточность каких именно факторов свертываемости наблюдается.
- Определение уровня гомоцистеина, позволяющее диагностировать гипергомоцистеинемию.
- Определение содержания тромбомодулина, активности фактора Виллебранда, а также концентрации эндотелина-1, необходимые для получения информации о возможном поражении сосудов вследствие тромбофилии.
- Генетическое исследование полиморфизма генов тех факторов, которые участвуют в гемостазе.
Наследственная тромбофилия и беременность
Связь между передаваемыми по наследству тромбофилиями и осложнениями беременности, например, преэклампсией, эклампсией, самопроизвольным прерыванием беременности, задержкой развития плода, отслойкой плаценты до сих пор остается противоречивой4. И тем не менее, некоторые акушерские осложнения все же вызваны тромбофилиями.
Так, ряд исследований свидетельствует о возможной связи между потерей плода во втором и третьем триместрах беременности с наличием наследственной тромбофилии6. Это объясняется нарушением кровообращения в сосудах плаценты вследствие формирования в ней тромбов. Однако важно подчеркнуть, что самопроизвольные потери плода на ранних сроках беременности (в первом триместре) чаще не имеют отношения к тромбофилии.
Однако важно подчеркнуть, что самопроизвольные потери плода на ранних сроках беременности (в первом триместре) чаще не имеют отношения к тромбофилии.
Тем не менее, наличие тромбофилии, которая заложена генетически, у беременных женщин может повышать риск венозных тромбоэмболических осложнений5. Даже в норме, у здоровых женщин во время беременности свертывающая система крови активируется: наблюдается физиологическая гиперкоагуляция (повышение свертываемости крови), также имеется венозный застой. Однако, у беременных женщин, страдающих тромбофилией, коагуляция еще более увеличивается, что и может приводить к повышению риска тромбообразования, прежде всего в венозных сосудах7.
Формированию венозного застоя у будущих мам может способствовать и снижение физической активности, например, при некоторых осложнениях беременности, а также после кесарева сечения 7.
Диагностика тромбофилии при беременности затруднена, поскольку ряд показателей повышается как при патологии, так и у здоровых женщин, причем чем больше срок беременности, тем выше риск тромбоза. Чтобы поставить диагноз, прибегают к генетическому исследованию, которое обычно проводится по рекомендации лечащего врача при наличии определенных показаний, например, при наличии перенесенного венозного тромбоза нижних конечностей или наличии венозных тромбозов у близких родственников.
Профилактика тромбозов
Профилактика тромбозов при тромбофилии в большинстве случаев основана на изменении образа жизни. Пациентам рекомендуют избегать тяжелых физических нагрузок и травм, при этом сохраняя прежний объем физической активности. Важная мера профилактики — ношение компрессионного трикотажа. Врачи всегда принимают решение о профилактических мерах строго индивидуально после оценки тромботического риска.
Список литературы
- Kyrle P.A., Rosendaal F.R., Eichinger S. Risk assessment for recurrent venous thrombosis //Elsevier.2010;376(9757):2032–2039.
- Khan S., Dickerman J.D. Hereditary thrombophilia. // Thromb. J. BioMed Central. 2006;4:15.
- Васильев С.А., Виноградов В.Л., и соавт. Тромбозы и тромбофилии: классификация, диагностика, лечение, профилактика // РМЖ — 2013.- № 17. — С.896.
- Robertson L. et al. Thrombophilia in pregnancy: a systematic review // Br. J. Haematol. Blackwell Science Ltd.2006;132(2):171–196.
- Sibai B.M., How H.Y., Stella C.L. Thrombophilia in pregnancy: Whom to screen, when to treat. 2007;19(1):50–64.
- Battinelli E.M., Marshall A., Connors J.M. The role of thrombophilia in pregnancy. // Thrombosis. Hindawi.2013; 2013:516420.
- Андрияшкин А.В. Российские клинические рекомендации по диагностике, лечению и профилактике венозных тромбоэмболических осложнений // Флебология — 2015. –Т. 9. – № 2. –С.1–52.
SARU.ENO.19.03.0436
Сдать анализ на гены системы гемостаза (с описанием результатов врачом- генетиком)
Метод определения
Real-time-PCR.
Исследуемый материал
Цельная кровь (с ЭДТА)
Доступен выезд на дом
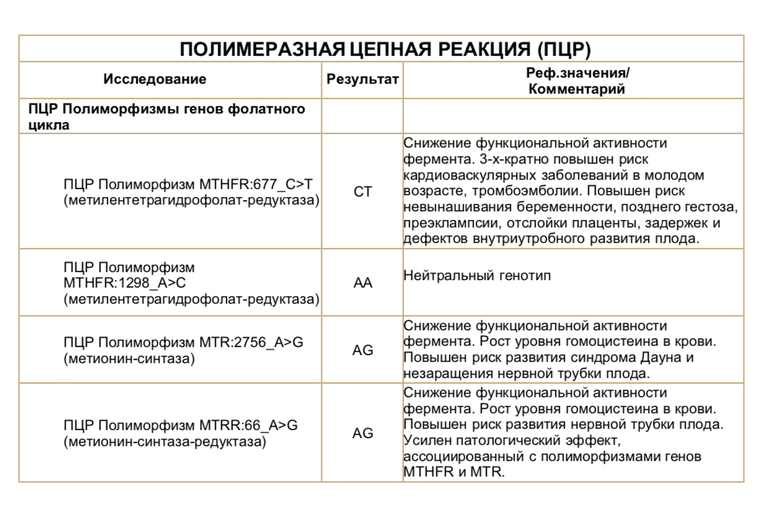
Расширенное исследование генов системы гемостаза: F2, F5, MTHFR, MTR, MTRR, F13, FGB, ITGA2, ITGВ3, F7, PAI-1
Комплексное исследование генетических факторов риска развития нарушений в системе свертывания крови и фолатном цикле.
Различные изменения в генах системы гемостаза и цикла обмена фолатов предрасполагают к развитию большого числа патологических состояний: инфаркты, инсульты, тромбоэмболии, кровотечения, патология беременности и родов, осложнения послеоперационного периода и т.д.
Профиль включает в себя исследование основных полиморфизмов в генах системы гемостаза и фолатного цикла:
- F2 c.*97G>A (20210 G>A; rs1799963),
- F5 c.1601G>A (Arg534Gln; 1691 G>A; rs6025),
- MTHFR c.665C>T (Ala222Val; 677 C>T; rs1801133),
- MTHFR c.1286A>C (Glu429Ala; 1298 A>C; rs1801131),
- MTR c.2756A>G (Asp919Gly; rs1805087),
- MTRR c.66A>G (Ile22Met; rs1801394),
- F13 с.103G>T (I63Т; rs5985),
- FGB c.-467G>A (-455 G>А; rs1800790),
- ITGA2 c.759C>T (Phe253Phe, 807 C>T; rs1126643),
- ITGB3 c.176T>C (Leu59Pro; 1565 T>C; rs5918),
- F7 c.1238G>A (Arg353Gln; 10976 G>A; rs6046),
- PAI-1 (SERPINE1) –675 5G>4G (rs1799889).
Ген F2 кодирует аминокислотную последовательность белка протромбина. Полиморфизм F2 c.*97G>A приводит к повышенной экспрессии гена. Клинически неблагоприятный вариант полиморфизма (c.*97A) наследуется по аутосомно-доминантному типу. Наличие полиморфизма F2 c.*97G>A в гомозиготной или гетерозиготной форме значительно (в 3 и более раз, а на фоне курения — в 40 и более раз) увеличивает риск возникновения венозных тромбозов, в том числе тромбозов сосудов мозга и сердца, особенно в молодом возрасте. У пациентов-носителей данного полиморфизма повышен риск развития тромбоэмболий после хирургических вмешательств. Приём оральных контрацептивов у данной группы лиц также увеличивает риск тромбозов (относительный риск развития тромбофилии и венозной тромбоэмболии у гетерозиготных носительниц полиморфизма c.*97G>A возрастает в 16 раз).
Ген F5 кодирует аминокислотную последовательность белка проакцелерина — коагуляционного фактора 5. Нуклеотидная замена c.1601G>A («мутация Лейден») приводит к аминокислотной замене аргинина на глутамин в позиции 534, что придает устойчивость активной форме проакцелерина. Клинически это проявляется рецидивирующими венозными тромбозами и тромбоэмболиями. Наличие полиморфизма в гомозиготной или гетерозиготной форме значительно (в 3 и более раз, а на фоне заместительной гормонотерапии или приема оральных контрацептивов — в 30 и более раз) увеличивает риск венозных тромбозов. Риск инфаркта миокарда увеличивается в 2 и более раз, риск развития патологии беременности (прерывание беременности, преэклампсия, хроническая плацентарная недостаточность и синдром задержки роста плода) увеличивается в 3 и более раз.
Также, пациенты, являющиеся одновременно носителями полиморфизма c.*97G>A гена протромбина и «мутации Лейден», еще в большей степени подвержены риску развития тромбозов и тромбоэмболий.
Ген MTHFR кодирует аминокислотную последовательность фермента метилентетрагидрофолатредуктазы, играющего ключевую роль в метаболизме фолиевой кислоты. Полиморфизм c.665C>T гена MTHFR связан с заменой нуклеотида цитозина (С) на тимин (Т), что приводит к аминокислотной замене аланина на валин в позиции 222. Вариант c.665Т связан с четырьмя группами мультифакториальных заболеваний: сердечно-сосудистыми, дефектами развития плода, колоректальной аденомой и раком молочной железы и яичников. У женщин с генотипом c.665Т/Т дефицит фолиевой кислоты во время беременности может приводить к порокам развития плода, в том числе незаращению нервной трубки. Неблагоприятное воздействие варианта c.665Т- зависит от внешних факторов: низкого содержания в пище фолатов, курения, приема алкоголя. Сочетание генотипа c.665Т/Т и папилломавирусной инфекции увеличивает риск цервикальной дисплазии. Назначение препаратов фолиевой кислоты может значительно снизить негативное влияние данного варианта полиморфизма.
Полиморфизм MTHFR c.1286A>C связан с точечной заменой нуклеотида аденина (А) на цитозин (С), что приводит к замене аминокислотного остатка глутаминовой кислоты на аланин в позиции 429, относящейся к регулирующей области молекулы фермента. При наличии данного полиморфизма отмечается снижение активности фермента MTHFR. Это снижение обычно не сопровождается изменением уровня гомоцистеина в плазме крови у носителей дикого варианта полиморфизма c.665C>T, однако сочетание аллельного варианта* c.1286C с аллелем c.665T приводит к снижению уровня фолиевой кислоты и соответствует по своему эффекту гомозиготному состоянию MTHFR c.665Т/T. При этом риск развития дефектов нервной трубки повышается в 2 раза. Жизнеспособность плодов, имеющих одновременно оба полиморфных варианта, также снижена.
Ген MTR кодирует аминокислотную последовательность фермента метионин синтазы. Полиморфизм c.2756A>G связан с аминокислотной заменой (аспарагиновой кислоты на глицин) в молекуле фермента. В результате этой замены функциональная активность фермента изменяется, что приводит к повышению риска формирования пороков развития у плода. Влияние полиморфизма усугубляется повышенным уровнем гомоцистеина.
Ген MTRR кодирует аминокислотную последовательность фермента редуктазы метионинсинтазы. Полиморфизм c.66A>G связан с аминокислотной заменой в молекуле фермента. В результате этой замены функциональная активность фермента снижается, что приводит к повышению риска развития дефектов нервной трубки у плода. Влияние полиморфизма усугубляется дефицитом витамина В12. При сочетании полиморфизма c.66A>G гена MTRR с полиморфизмом c.665C>T в гене MTHFR риск spina bifida увеличивается. Полиморфизм c.66A>G гена MTRR усиливает гипергомоцистеинемию, вызываемую полиморфизмом c.665C>T в гене MTHFR.
Ген фибриназы (F13) кодирует синтез трансглютаминазы, участвующей в стабилизации фибринового сгустка и в формировании соединительной ткани. Аллельные варианты с.103G/Т и с.103Т/Т приводят к снижению уровня трансглютаминазы с образованием сетчатой структуры фибрина с более тонкими волокнами, меньшими порами, и изменением характеристик проникновения, которое в сочетании с другими факторами риска ассоциируется с возможным риском внутричерепных кровоизлияний и кровотечений из внутренних органов, а также привычным невынашиванием беременности. При этом аллельный вариант с.103Т может выступать в роли протективного фактора в отношении инфаркта миокарда и венозных тромбозов.
Ген FGB кодирует β-цепь фибриногена, являющегося предшественником фибрина. Аллельный вариант c.-467А обусловливает усиленную транскрипцию гена и может приводить к увеличению уровня фибриногена в крови и повышению вероятности образования тромбов при наличии дополнительных факторов риска. Гетерозиготный вариант c.-467G/А связывают с повышенным риском ишемического инсульта и лакунарными инфарктами церебральных сосудов. Гомозиготный вариант c.-467A/А связывают с повышенным риском инфаркта миокарда.
Ген гликопротеина Gp1a (ITGA2) кодирует синтез альфа-2-субъединицы интегринов – специализированных рецепторов тромбоцитов. Аллельный вариант c.759Т вызывает изменение первичной структуры субъединицы и свойств рецепторов. При гетерозиготном (c.759C/T) варианте отмечается увеличение скорости адгезии тромбоцитов к коллагену I типа, что может приводить к повышенному риску тромбофилии, инфаркта миокарда и других сердечно-сосудистых заболеваний. Аллельный вариант c.759Т связывают со случаями резистентности к аспирину. Помимо этого, при гомозиготном (c.759Т/T) варианте значительно увеличивается количество рецепторов на поверхности тромбоцитов. В совокупности, при гомозиготном варианте данного полиморфизма значительно повышен риск тромбофилии, инфаркта миокарда и развития других острых эпизодов тромбообразования в возрасте до 50 лет, даже по сравнению с гетерозиготным вариантом.
Ген гликопротеина Gp3a (ITGB3) кодирует синтез бета-3 цепи интегринового комплекса GP2b\3a, участвующего в разнообразных межклеточных взаимодействиях (адгезии и сигнализации).
Аллельный вариант c.176С (гетерозигота c.176T/C) обусловливает повышенную адгезию тромбоцитов и может приводить к увеличению риска развития острого коронарного синдрома, а также связан с синдромом привычного невынашивания беременности. Гомозиготный вариант c.176С/C обусловливает повышенную адгезию тромбоцитов и может приводить к значительному увеличению риска развития острого коронарного синдрома в возрасте до 50 лет. У лиц с полиморфными аллельными вариантами часто отмечается пониженная эффективность аспирина.
Аллельный вариант c.1238A (гетерозигота c.1238G/A и гомозигота c.1238А/A) гена F7 приводит к понижению экспрессии гена и снижению уровня фактора 7 в крови, рассматривается как протективный маркёр в отношении развития тромбозов и инфаркта миокарда.
Ген ингибитора активатора плазминогена (PAI-1) кодирует белок-антагонист тканевого и урокиназного активатора плазминогена. Преобладающим в популяции вариантом исследуемого полиморфизма является гетерозиготный вариант -675 5G/4G. В связи с этим данный полиморфизм самостоятельного диагностического значения не имеет, эффект возможно оценить в сочетании с другими факторами предрасполагающими к развитию патологии (например в сочетании с FGB c.-467A). Аллельный вариант -675 4G сопровождается большей активностью гена, чем -675 5G, что обусловливает более высокую концентрацию PAI-1 и уменьшение активности противосвёртывающей системы. Гомозигота -675 4G/4G ассоциирована с повышением риска тромбообразования, преэклампсии, нарушением функции плаценты и самопроизвольного прерывания беременности.
*Примечание: иногда в научной литературе при описании однонуклеотидных замен, характерных для генных полиморфизмов, встречается термин «мутантный аллель». Это терминологическая неточность, так как в классической генетике термин «мутантный аллель» традиционно рассматривается как синоним термина «мутация». При мутациях, как известно, изменение структуры гена приводит к образованию (экспрессии) нефункциональных белков и к неизбежному развитию наследственного заболевания. При полиморфизмах изменение в структуре гена приводит лишь к появлению белков с немного изменёнными физико-химическими свойствами. Такие изменения, как известно, проявляют себя при воздействии на организм различных факторов внешней среды или при изменении функционального состояния организма человека. И только в таких ситуациях функционирование белков со структурными особенностями может, либо способствовать ускорению развития заболевания, либо, напротив, тормозить формирование патологических процессов. Поэтому, на наш взгляд, для разграничения изменений в генах столь очень похожих структурно, но приводящих к несоизмеримо разным последствиям для организма, корректнее в отношении генных полиморфизмов применять понятие «аллельный вариант гена», а не «мутантный аллель».
Краткий словарь генетических терминов
Краткий словарь основных понятий и терминов, использующихся в генетике
Для понимания того, с чем работает наша компания и зачем эта работа нужна, какие результаты мы получаем и что они вам расскажут, можно прийти на консультацию к специалистам ЦГРМ «ГЕНЕТИКО». А для того, чтобы Вы не забыли, о чем был разговор, и не утонули в море новой информации, мы составили для Вас небольшой словарик основных понятий и терминов, использующихся в генетике.
Основным положением биологической науки является то, что клетка – это самое маленькое из возможных проявление жизни и что новая клетка может появиться только от уже существующей и никак не может возникнуть сама по себе. Конечно, это приводит к большому количеству вопросов о том, как зародилась жизнь и каким образом могла сформироваться самая первая клетка. Но для удобства будем считать обозначенные положения верными в современной реальности планеты Земля, где мы живем. Несмотря на невообразимо огромное разнообразие живых существ, все они состоят из клеток. И у всех клеток есть схожие черты, которые обусловлены самыми простыми жизненными необходимостями. Во-первых, клетка должна как-то отделяться от внешнего пространства – для этого есть специальная оболочка.
Во-вторых, клетка должна питаться – для этого есть разные системы, способные преобразовать энергию света или химических связей в необходимые для жизни вещества и удобную для использования энергию. И еще клетка умеет размножаться. Для выполнения всех этих функций необходимы механизмы, основу которых составляют белки и РНК. А вот инструкция, как эти молекулы должны выглядеть и работать, хранится в специальном отсеке клетки – ядре – в виде ДНК. Ошибки в этой инструкции, которая разрабатывалась миллионы лет, приводят к смерти клетки. А в многоклеточном организме, таком, как у человека, например, клетки взаимодействуют друг с другом, поэтому нарушение в работе одной или нескольких клеток может привести не к смерти всего организма, а к нарушениям его работы – заболеваниям. Также необходимо помнить, что человеческий организм огромная система, ансамбль миллионов разнообразных маленьких организмов, которые выросли из одной единственной клетки – зиготы – результата слияния яйцеклетки и сперматозоида.
ДНК – ДезоксиРибонуклиновая Кислота – полимер, то есть молекула с большим количеством последовательно повторяющихся структурных элементов, который несет всю информацию о генах и белках, необходимых для жизни всего организма. ДНК является картотекой, библиотекой и матрицей, с которой считывается информация в определенной последовательности и определенных условиях, разъяснения о которых записаны как в самой ДНК, так и с помощью различных дополнительных модификаций этой молекулы. Каждой хромосоме соответствует 1 молекула ДНК. Структурными блоками этого полимера являются дезоксирибонуклеотиды (=нуклеотиды), которые бывают 4х видов (А, Т, Г, Ц).
Последовательность ДНК – это то, в каком порядке в молекуле ДНК идут ее структурные элементы – нуклеотиды. Таким образом, генетической информацией является именно последовательность ДНК, а молекула ДНК является ее физическим носителем.
Хромосома – это молекула ДНК, специальным образом обернутая различными белками, которые помогают управляться с такой длинной молекулой, чтобы она не порвалась, не перепуталась с другими ДНК-молекулами и была физически доступна для белков, осуществляющих работу всего генетического аппарата.
РНК –РибоНуклиновая Кислота – полимер, который выполняет функциональную роль переносчика информации, то есть копии, которая делается с ДНК и используется для создания функциональных молекул: специальных РНК или белков. Специальные молекулы РНК могут не являться матрицами, на базе которых синтезируется белок, а сами выполняют структурные, ферментативные или транспортные функции. Главное, что последовательность структурных блоков в молекуле РНК всегда определена последовательностью ДНК соответствующего участка.
Белок – основная функциональная единица живой клетки с самым широчайшим спектром функций и возможностей. Как ДНК и РНК, является полимером, однако имеет химически иные структурные блоки – аминокислоты. Их последовательность, с одной стороны, напрямую зависит от соответствующей последовательности ДНК и может изменяться только в ограниченных и предусмотренных в ДНК инструкций, с другой стороны является основой структуры, в том числе пространственной, возможностей и функции белков разных типов.
Ген – определение гена включает два аспекта: теоретический и физический. Теоретически, то есть умозрительно, геном называют последовательность ДНК (слово, записанное на языке генетики), обладающее определенными свойствами. Как и слово в языке, ген является основой наследственной информации, в то время как различные другие структуры можно отнести к знакам препинания или вспомогательным элементам. Ген является подробной инструкцией для синтеза белка или специфической РНК, которую он кодирует. Причем эта инструкция описывает не только последовательность молекул, но и то в каких условиях и как они должны работать и выполнять свои функции. С физической, то есть материальной, точки зрения, ген – это часть молекулы ДНК с определенными структурными элементами. Как внутри слова есть приставка, корень, суффикс и окончание, позволяющие слову адаптироваться для каждой конкретной фразы, так и у гена есть промотор, экзоны и интроны. Первый обозначает начало гена, экзоны – это ключевая информация о последовательности РНК или белка, а интроны необходимы для регуляции и тонкой настройки работы гена в условиях разных тканей, органов и изменяющейся окружающей среды.
Экспрессия гена – это эффективность работы гена, так как для его функционирования недостаточно его наличия в геноме – с него должна считываться информация. Именно то, как часто и в каком объеме считывается информация с гена, выражают термином экспрессия.
Локус – участок молекулы ДНК, содержащий различный структурные элементы, в том числе один или несколько генов.
Геном– это последовательность всех молекул ДНК организма. Важно помнить, что в каждой клетке одного организма в норме содержатся одинаковые по количеству и последовательностям молекулы ДНК, а различается экспрессия конкретных генов.
Экзом – это последовательности ДНК экзомных участков генов, то есть так называемая основная кодирующая составляющая. Это то, с чем работает организм, в то время как остальная часть генома объясняет, как работать и в каких условиях как применять и настраивать кодирующую часть генома.
Мутация – изменение последовательности ДНК по сравнению другими клетками организма или другими представителями вида. Мутации могут возникать как из-за воздействия внешних неблагоприятных условий, так и из-за того, что наши ферменты работают пусть с редкими, но ошибками. Так как происходит физическое изменение в носителе информации – ДНК, такое изменение может передаваться из поколения в поколение.
Частота мутаций — относительное значение, показывающее у какой доли людей в геноме есть конкретная мутация. Частоту мутации можно рассчитать, как среднюю для всех людей, так и отдельно по расовым или национальным, или любы другим группам. В медицинской генетике под мутацией подразумевают изменение ДНК, которое может быть связано с каким-то заболеванием, и противопоставляют ее полиморфизму. Хотя по общей логике полиморфизм – это частный случай мутации.
Полиморфизм – нейтральная, а точнее безвредная, мутация, которая сравнительно часто встречается у какой-то группы организмов одного вида. Некоторые мутации встречаются часто у всех людей, некоторые – только среди представителей определенных рас или народностей.
Аллель – вариант последовательности гена в разном виде: от различия в одной букве последовательности до отсутствия целого куска последовательности или вставке лишнего. Эти различия возникают из-за мутации, которая могла произойти у далекого предка и передаться потомству через поколения. Таким образом, каждый ген у отдельного человека может быть представлен конкретным вариантом – аллелем. Для понимания аллелизма необходимо объяснить, что, например, различия в цвете глаз, волос, росте, чувствительности к алкоголю объясняются именно разными аллельными состояниями соответствующих генов.
Генотип – это все гены конкретной особи с указанием аллельного состояния каждого гена и наличия/отсутствия мутаций в межгенных участках ДНК.
Доминантный аллель. В геноме человека содержится по 2 копии каждой хромосомы. Это означает, что в каждом геноме есть две очень похожие по длине и последовательности генов молекулы ДНК, которые отличаются аллельными состояниями генов и мутациями/полиморфизмами в межгенных участках этих молекул ДНК. Из этого следует, что и каждый ген представлен в геноме 2 копиями, каждая из которых может быть определенным вариантом (аллелем) этого гена. Доминантным аллелем называется тот, одной копии которого достаточно для проявления его особенностей. То есть если хотя бы на одной из хромосом ген находится в состоянии доминантного аллеля, то ген будет работать по тому варианту, который описывается именно этим аллелем. Важно, что так как у одного гена может быть более двух вариантов (аллелей), то и доминантность аллеля определяется по отношению к каждому из вариантов, хотя есть и те, которые доминантны по сравнению со всеми другими. Встречаются варианты с одинаковой предпочтительностью для работы, тогда проявляется совместное влияние этих вариантов.
Рецессивный аллель – по аналогии с доминантным аллелем, это такое состояние гена, которое наименее предпочтительно для проявления. Поэтому если в геноме есть другая копия гена, доминантная, то задавать темп работы гена будет именно она, но если и вторая копия гена представлена рецессивным аллелем, то будет работать этот, хотя менее предпочтительный, но в такой ситуации единственно имеющийся вариант. Хотя в большинстве случаев связанные с возникновением заболевания аллели рецессивны, это вредность/полезность не является единственным определяющим фактором рецессивности/доминантности аллеля.
Гомозигота. Гомозиготой по определенной мутации/полиморфизму/аллелю называют такую клетку или организм, в генотипе которой/которого обе копии гена на двух хромосомах представлены одним вариантом, то есть не отличаются по этой мутации/полиморфизму/аллелю.
Гетерозигота. Гетерозиготой по определенной мутации/полиморфизму/аллелю называют такую клетку или организм, в генотипе которой/которого две копии гена на двух хромосомах представлены разными вариантами, то есть отличаются по этой мутации/полиморфизму/аллелю.
Секвенирование – это группа методов, позволяющая узнать последовательность нуклеотидов в молекуле ДНК. Этот метод обладает некоторыми особенностями. Во-первых, пока что ни один способ секвенирования не позволяет прочитать всю последовательность одной хромосомы, чтение идет сравнительно небольшими отрезка от 50 до несколько тысяч нуклеотидов. Во-вторых, почти все методы устроены так, что из кусочка ДНК делается много одинаковых и читаются они все. Эта особенность проявляется в таком параметре секвенирования, как глубина секвенирования, обозначаемая 10Х, 20Х, 50Х. Чем больше это значение, тем больше раз прочитан один и тот же кусок молекулы, тем точнее можно выявить ошибки секвенирования и особенности участка, например, его гетерозиготность по какой-либо мутации/полиморфизму.
Гаплотип — совокупность состояний/вариантов определенных локусов, которые расположены на одной хромосоме, и вследствие структурных особенностей эти состояния всегда наследуются вместе. То есть, например, если в одном локусе (1) гаплотипа имеется мутация (1А), а в другом (2) имеется уже другая мутация (2M), то именно в таком составе они будут наследоваться (1А2М), а смешанных вариантов (1B2M или 1A2N) не бывает или они относятся к другому гаплотипу.
Гаплогруппа — совокупность особей, имеющих сходный гаплотип по определенным локусам, которые задаются в соответствии с тем, какую задачу нужно решить, определяя гаплогруппу
Митохондриальная ДНК. Если разбираться подробнее и глубже, то генетическая информация одного человека находится не только в 46 хромосомах, располагающихся в специальном отсеке клетки – ядре, но и в клеточных органах митохондриях. У митохондрий в клетке своя задача – преобразовывать энергию, заключенную в химической связи определенных атомов, в более удобную для клетки, то есть они готовят эффективные питательные запасы из разного сырья. Митохондрии довольно сложны, их оболочка хитро устроена, чтобы опасные побочные продукты готовки не могли попасть в остальную часть клетки, поэтому все время таскать туда нужные для их работы белки не слишком продуктивно. Таким образом, у них есть своя ДНК, которая несет информацию о разных особенных белках и РНК, которые нужны именно для работы митохондрии. Такую ДНК называют митохондриальной и она является неотъемлемой и обязательной частью нашего генотипа. Передается она только от мамы, так как сперматозоид для возможности быстро перемещаться и долго оставаться живым несет самый минимум необходимой генетической информации – 23 хромосомы. А вот яйцеклетка, которой для выполнения основной функции не нужно находится в агрессивной окружающей среде, может позволить себе бОльшую массу и дополнительные запасы в виде готовых к работе станций приготовления питания – митохондрий и заранее синтезированных белков и РНК.
Гены половой дифференцировки – группа генов, играющая ведущую роль в определении будет эмбрион развиваться как девочка или как мальчик. В геноме человека основой проявления мужских или женских половых признаков является наличие/отсутствие половой хромосомы Y, а именно особо локуса этой хромосомы – SRY (Sex-determining Region on the Y chromosome). Важно отметить, что нарушения в этом локусе могут приводить не к внешним проявлениям, а к сниженной репродуктивной способности мужчины или ее полному отсутствию. Процесс дифференцировки пола у человека можно представить тремя стадиями: 1) какой набор хромосом получается при слиянии яйцеклетки (всегда несет хромосому X) и сперматозоида (с хромосомой X или Y), 2) формирование женских или мужских половых органов в зависимости от работы генов локуса SRY, 3) развитие вторичных половых органов в соответствии с типом половых органов. Нарушения на разных этапах приводят к разным проявлениям и разным заболеваниям.
Локус AZF – это участок Y-хромосомы, на котором располагаются так называемые факторы азооспермии (AZF — AZoospermia Factors). Это особые участки, которые названы так, потому что если какой-то из них отсутствует из-за мутации, то развивается азооспермия (отсутствие сперматозоидов) или олигозооспермия (малое количество сперматозоидов). Всего обнаружено три таких фактора AZFa, AZFb и AZFc. В норме наличие всех трех является минимальным необходимым условием нормального формирования сперматозоидов. Если в геноме отсутствует один из AZFa и AZFb или оба, то нарушается созревание сперматозоидов и, как следствие, полностью отсутствует репродуктивная функция. При отсутствии локуса AZFc нарушения могут быть не столь сильными, поэтому деторождение остается возможным в некоторых случаях.
Хромосомные аномалии – это крупные мутации, которые связаны с изменением последовательности ДНК не в рамках отдельного гена или нескольких, а в масштабе хромосомы или генома. Например, отсутствие (делеция) большой части или всей хромосомы, лишняя хромосома, или часть одной хромосомы соединена с частью другой хромосомы и т.д.
Наследственное заболевание – это заболевание, вызванное нарушениями в геноме, то есть мутациями, которые либо мешают формированию нормального белка (так как ген – инструкция по его построению – поврежден), либо изменяют регуляцию, то есть условия, когда, в каком месте или с кем такой белок или ген должен работать.
Моногенное заболевание – это наследственное заболевание, вызванное мутацией в одном только в одном гене. Несмотря на то, что все остальные почти 30000 генов могут быть в порядке, изменение последовательности ДНК в этом гене вызывает нарушения функционирования всего организма.
Хромосомное заболевание – наследственное заболевание, вызванное хромосомными аномалиями.
Носительство мутации – это состояние гетерозиготы по аллелю, обладающему какими-то негативными клиническими проявлениями, если он находится в геноме в виде гомозиготы.
Пробанд – человек, с которого начинается составление генеалогического дерева (родословной). Обычно пробанд – это носитель или пациент с наследственным заболеванием, проявление которого и вызвало необходимость генеалогического анализа.
Сиблинг – в генетике таким термином обозначают потомков одних родителей, то есть братьев и сестер, но не близнецов.
Автор: Жикривецкая Светлана
Биолог-исследователь
Цены на услуги – Генетический паспорт
Обратите внимание, что данная страница с ценами — это калькулятор. Вы можете выбрать в выпадающем списке ниже интересующий вас комплекс, нажимать на блоки с ценами, чтобы узнать итоговую стоимость. Мы не запоминаем ваш выбор, эти данные никуда не отправляются и нигде не хранятся! Пожалуйста, учитывайте это, когда будете записываться на приём.
Загрузить прейскурант в виде документа pdf можно по этой ссылке .
Основной комплекс 0
Дополнительный комлекс 0
Полный комплекс 0
Итоговая стоимость выбранных генов составляет
171.00 BYN
Показать цены для нерезидентов
Уникальная технология количественной оценки генетического риска невынашивания беременности (обновлено)
Внимание. Мы снижаем стоимость исследования – с 01.11.2021 до 31.12.2021 года действует акция, по которой цена тестирования комплекса из 25 генов, ассоциированного с невынашиванием беременности, СНИЖЕНА НА 25%.
Полный текст читайте в разделе сайта «Новости»
Цены на сайте указаны без скдики.
Генная картина – это уникальный генетический портрет человека.
Художественное переосмысление научных достижений в генетике позволяет создавать генетический портрет человека. Картина представляет собой необычное изображение, основу которого составляют гены конкретного человека. И сколько бы тысяч лет ни прошло, на Земле не появится вторая такая картина. Потому что ДНК-код, зашифрованный в наших генах, уникален и никогда не повторяется.
Генные картины – это возможность сделать интерьер своего дома по-настоящему уникальным, это не только изысканное и новаторское украшение интерьера, но еще и прекрасно оформленное наглядное доказательство нашей природной уникальности, которое создает в доме неповторимую атмосферу и придает ему незабываемые индивидуальные черты. Генная картина.
Этапы создания:
- ДНК, выделенная из слюны либо капли крови человека, подвергается полимеразной цепной реакции, в ходе которой определенные участки ДНК умножаются (копируются) в миллион раз. Далее раствор, насыщенный миллионами копий фрагментов ДНК наносится на специальный гель, к которому подается электрическое напряжение. Под воздействием электрического поля исследуемые фрагменты ДНК перемещаются на определенные расстояния, пропорциональные их массе, создавая неповторимый графический узор;
- Изображение наносится методом цифровой печати на бумагу, стекло либо создается художником на холсте. Заказчик сам выбирает материал, цветовое решение и размер картины.
Вместе с генной картиной выдается сертификат, подтверждающий достоверность и уникальность данного генетического портрета.
Картина представляет собой необычное изображение, основу которого составляют гены конкретного человека, и отображает его генетическую уникальность. Прекрасно оформленная генная картина создает в доме неповторимую атмосферу и придает ему незабываемые индивидуальные черты. Вместе с картиной выдаётся сертификат, подтверждающий достоверность и уникальность данного генетического портрета.
Стоимость анализа для резидентов Республики Беларусь составляет 367.60 BYN. Для нерезидентов – 460.20 BYN. Стоимость указана без разработки оригинал-макета генной картины и без печатных работ.
Паспорт здоровья содержит в себе результаты генетического анализа по всем полиморфизмам генов, исследуемых в лаборатории.
Отличная идея!
Получите второй генетический паспорт по предрасположенности к ССЗ (сердечно-сосудистым заболеваниям) всего за BYN! Гены, ответственные за невынашивание, во время не беременности никуда не «исчезают». И их неблагоприятные эффекты можно рассматривать как риск развития сердечно-сосудистых заболеваний (инсульты, инфаркты и др.). Получить второй генетический паспорт – это отличная идея.
Архитектура полиморфизмов в геноме человека выявляет функционально важные и положительно выбранные варианты в генах иммунного ответа и переносчиков лекарств | Human Genomics
Коллинз Ф.С., Брукс Л.Д., Чакраварти А. Ресурс по открытию полиморфизма ДНК для исследования генетической изменчивости человека. Genome Res. 1998. 8: 1229–31.
CAS
Статья
Google ученый
Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al.Общие регуляторные вариации влияют на экспрессию генов в зависимости от типа клетки. Наука. 2009; 325: 1246–50.
CAS
Статья
Google ученый
Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET. Генетика транскриптома с использованием секвенирования второго поколения в популяции европеоидной расы. Природа. 2010; 464: 773–7.
CAS
Статья
Google ученый
Майерс А.Дж., Гиббс Дж. Р., Вебстер Дж. А., Рорер К., Чжао А., Марлоу Л., Калим М., Леунг Д., Брайден Л., Нат П. и др. Обзор генетической экспрессии генов коры головного мозга человека. Нат Жене. 2007; 39: 1494–9.
CAS
Статья
Google ученый
Пикрелл Дж. К., Мариони Дж. К., Пай А. А., Дегнер Дж. Ф., Энгельгардт Б. Э., Нкадори Е., Вейриерас Дж. Б., Стивенс М., Гилад Ю., Притчард Дж. К.. Понимание механизмов, лежащих в основе вариабельности экспрессии генов человека, с помощью секвенирования РНК.Природа. 2010; 464: 768–72.
CAS
Статья
Google ученый
Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. Картирование генетической архитектуры экспрессии генов в печени человека. PLoS Biol. 2008; 6: e107.
Артикул
Google ученый
Stranger BE, Forrest MS, Clark AG, Minichiello MJ, Deutsch S, Lyle R, Hunt S, Kahl B., Antonarakis SE, Tavare S, et al.Полногеномные ассоциации вариаций экспрессии генов у людей. PLoS Genet. 2005; 1: e78.
Артикул
Google ученый
Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, et al. Популяционная геномика экспрессии генов человека. Нат Жене. 2007; 39: 1217–24.
CAS
Статья
Google ученый
Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, Pritchard JK.Картирование экспрессионных QTL с высоким разрешением дает представление о регуляции генов человека. PLoS Genet. 2008; 4: e1000214.
Артикул
Google ученый
Целлер Т., Вайлд П., Шимчак С., Ротиваль М., Шиллер А., Кастань Р., Мауш С., Жермен М., Лакнер К., Россманн Х. и др. Генетика и не только — транскриптом человеческих моноцитов и восприимчивость к болезням. PLoS One. 2010; 5: e10693.
Артикул
Google ученый
Аджубей ИА, Шмидт С, Пешкин Л, Раменский В.Е., Герасимова А, Борк П, Кондрашов АС, Сюняев СР. Метод и сервер для предсказания повреждающих миссенс-мутаций. Нат методы. 2010; 7: 248–9.
CAS
Статья
Google ученый
Карчин Р., Диханс М., Келли Л., Томас Д. Д., Пипер У., Эсвар Н., Хаусслер Д., Сали А. LS-SNP: крупномасштабная аннотация кодирования несинонимичных SNP на основе нескольких источников информации. Биоинформатика.2005; 21: 2814–20.
CAS
Статья
Google ученый
Юэ П., Меламуд Э., Молт Дж. SNPs3D: ген-кандидат и отбор SNP для ассоциативных исследований. BMC Bioinformatics. 2006; 7: 166.
Артикул
Google ученый
Ng PC, Хеникофф С. Учет человеческих полиморфизмов, которые, по прогнозам, влияют на функцию белка. Genome Res. 2002; 12: 436–46.
CAS
Статья
Google ученый
Ли Б., Кришнан В.Г., Морт М.Э., Синь Ф., Камати К.К., Купер Д.Н., Муни С.Д., Радивояк П. Автоматизированный вывод молекулярных механизмов заболевания на основе аминокислотных замен. Биоинформатика. 2009; 25: 2744–50.
CAS
Статья
Google ученый
Thomas PD, Kejariwal A, Guo N, Mi H, Campbell MJ, Muruganujan A, Lazareva-Ulitsky B. Приложения для данных эволюции последовательности-функции белка: анализ экспрессии мРНК / белка и инструменты оценки кодирующих SNP.Nucleic Acids Res. 2006; 34: W645–50.
Артикул
Google ученый
Каприотти Э., Калабрезе Р., Касадио Р. Прогнозирование роста генетических заболеваний человека, связанных с точечными мутациями белков, с помощью опорных векторных машин и эволюционной информации. Биоинформатика. 2006; 22: 2729–34.
CAS
Статья
Google ученый
Масица Д.Л., Карчин Р.На пути к повышению клинической значимости методов in silico для прогнозирования патогенных миссенс-вариантов. PLoS Comput Biol. 2016; 12: e1004725.
Артикул
Google ученый
Петерсон Т.А., Даути Э., Канн М.Г. На пути к точной медицине: достижения в вычислительных подходах к анализу человеческих вариантов. J Mol Biol. 2013; 425: 4047–63.
CAS
Статья
Google ученый
Фабер К., Глаттинг К.Х., Мюллер П.Дж., Риш А., Хотц-Вагенблатт А. Прогнозирование в масштабе всего генома модифицирующих сплайсинг SNP в генах человека с использованием нового конвейера анализа, называемого AASsites. BMC Bioinformatics. 2011; 12 (Приложение 4): S2.
Артикул
Google ученый
Ян Дж.О., Ким В.Й., Бхак Дж. SsSNPTarget: полногеномная база данных однонуклеотидного полиморфизма сайтов сплайсинга. Hum Mutat. 2009; 30: E1010–20.
Артикул
Google ученый
Kim BC, Kim WY, Park D, Chung WH, Shin KS, Bhak J. SNP @ Promoter: база данных человеческих SNP (однонуклеотидных полиморфизмов) в предполагаемых промоторных областях. BMC Bioinformatics. 2008; 9 (Приложение 1): S2.
Артикул
Google ученый
Bao L, Zhou M, Wu L, Lu L, Goldowitz D, Williams RW, Cui Y. База данных PolymiRTS: связывание полиморфизмов в целевых сайтах микроРНК со сложными признаками. Nucleic Acids Res. 2007; 35: D51–4.
CAS
Статья
Google ученый
Hiard S, Charlier C, Coppieters W, Georges M, Baurain D. Patrocles: база данных полиморфной miRNA-опосредованной регуляции генов у позвоночных. Nucleic Acids Res. 2010; 38: D640–51.
CAS
Статья
Google ученый
Гонг Дж., Тонг И, Чжан Х.М., Ван К., Ху Т., Шань Дж., Сунь Дж., Го А.Ю. Полногеномная идентификация SNP в генах микроРНК и влияние SNP на связывание микроРНК-мишени и биогенез. Hum Mutat. 2012; 33: 254–63.
CAS
Статья
Google ученый
Ван Дж., Ронаги М., Чонг С.С., Ли К.Г. pfSNP: интегрированный потенциально функциональный ресурс SNP, который облегчает генерацию гипотез посредством синтеза знаний. Hum Mutat. 2011; 32: 19–24.
Артикул
Google ученый
Conde L, Vaquerizas JM, Santoyo J, Al-Shahrour F, Ruiz-Llorente S, Robledo M, Dopazo J.PupaSNP Finder: веб-инструмент для поиска SNP с предполагаемым эффектом на уровне транскрипции. Nucleic Acids Res. 2004; 32: W242–8.
CAS
Статья
Google ученый
Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Опрос карты SNP с высокой плотностью на предмет признаков естественного отбора. Genome Res. 2002; 12: 1805–14.
CAS
Статья
Google ученый
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, et al. Полногеномное обнаружение и характеристика положительного отбора в человеческих популяциях. Природа. 2007; 449: 913–8.
CAS
Статья
Google ученый
Баррейро Л.Б., Лаваль Дж., Куач Х., Патин Э., Кинтана-Мурси Л. Естественный отбор привел к дифференциации популяций современных людей. Нат Жене. 2008; 40: 340–5.
CAS
Статья
Google ученый
Чен К., Раевский Н. Естественный отбор на сайтах связывания микроРНК человека, выведенный из данных SNP. Нат Жене. 2006; 38: 1452–6.
CAS
Статья
Google ученый
Jha P, Lu D, Xu S. Естественный отбор и функциональные возможности некодирующих элементов человека, выявленные в результате анализа данных секвенирования следующего поколения.PLoS One. 2015; 10: e0129023.
Артикул
Google ученый
Энард Д., Мессер П.В., Петров Д.А. Общегеномные сигналы положительного отбора в эволюции человека. Genome Res. 2014; 24: 885–95.
CAS
Статья
Google ученый
Нильсен Р. Молекулярные признаки естественного отбора. Анну Рев Жене. 2005; 39: 197–218.
CAS
Статья
Google ученый
Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D. Ультраконсервированные элементы в геноме человека. Наука. 2004; 304: 1321–5.
CAS
Статья
Google ученый
Вандиедонк К., Найт Дж. К.. Главный комплекс гистосовместимости человека как парадигма в геномных исследованиях. Краткая функция геномной протеомики. 2009; 8: 379–94.
CAS
Статья
Google ученый
Зангер У.М., Шваб М. Ферменты цитохрома P450 в метаболизме лекарств: регуляция экспрессии генов, активности ферментов и влияние генетической изменчивости. Pharmacol Ther. 2013; 138: 103–41.
CAS
Статья
Google ученый
Бересфорд А.П. CYP1A1: друг или враг? Drug Metab Rev.1993; 25: 503–17.
CAS
Статья
Google ученый
Ван Б., Ян Л.П., Чжан ХЗ, Хуанг С.К., Бартлам М., Чжоу С.Ф.Новое понимание структурных характеристик и функциональной значимости фермента цитохрома P450 2D6 человека. Препарат Метаб Ред. 2009; 41: 573–643.
CAS
Статья
Google ученый
Лепешева Г.И., Ватерман М.Р. Стерол 14альфа-деметилаза цитохром P450 (CYP51), P450 во всех биологических царствах. Biochim Biophys Acta. 2007; 1770: 467–77.
CAS
Статья
Google ученый
Дин М., Ржецкий А., Алликметс Р. Суперсемейство переносчиков человеческих АТФ-связывающих кассет (АВС). Genome Res. 2001; 11: 1156–66.
CAS
Статья
Google ученый
Бейкер А., Диджей Кэрриер, Шедлер Т., Уотерхэм Х. Р., ван Рурмунд К. В., Теодулу, Флорида. Переносчики пероксисомальных ABC: функции и механизм. Biochem Soc Trans. 2015; 43: 959–65.
CAS
Статья
Google ученый
Абеллан Р., Мансего М.Л., Мартинес-Эрвас С., Мартин-Эскудеро Дж. К., Кармена Р., Реал Дж. Т., Редон Дж., Кастродеза-Санс Дж. Дж., Чавес Ф. Дж. Ассоциация выбранных однонуклеотидных полиморфизмов семейства генов ABC с липопротеинами после приема пищи: результаты популяционного исследования Hortega. Атеросклероз. 2010; 211: 203–9.
CAS
Статья
Google ученый
Akey JM. Построение геномных карт положительного отбора у людей: что нам делать дальше? Genome Res.2009; 19: 711–22.
CAS
Статья
Google ученый
Brion M, Sanchez-Salorio M, Corton M, de la Fuente M, Pazos B, Othman M, Swaroop A, Abecasis G, Sobrino B, Carracedo A, Испанская многоцентровая группа AMD. Исследование генетической ассоциации возрастной дегенерации желтого пятна у населения Испании. Acta Ophthalmol. 2011; 89: e12–22.
Артикул
Google ученый
Chu LW, Li Y, Li Z, Tang AY, Cheung BM, Leung RY, Yik PY, Jin DY, Song YQ. Новый интронный полиморфизм гена ABCA1 показывает риск спорадической болезни Альцгеймера у китайцев. Am J Med Genet B Neuropsychiatr Genet. 2007; 144B: 1007–13.
CAS
Статья
Google ученый
Джеймисон С.Е., де Рубе Л.А., Кортина-Борха М., Тан Х.К., Муи Э.Дж., Корделл Х.Дж., Кириситс М.Дж., Миллер Э.Н., Пикок С.С., Харгрейв А.С. и др. Генетические и эпигенетические факторы COL2A1 и ABCA4 влияют на клинический исход врожденного токсоплазмоза.PLoS One. 2008; 3: e2285.
Артикул
Google ученый
Джордан де Луна С., Эрреро Сервера М.Дж., Санчес Лазаро I, Альменар Бонет Л., Поведа Андрес Дж.Л., Алино Пеллисер С.Ф. Фармакогенетическое исследование генов ABCB1 и CYP3A5 в течение первого года после трансплантации сердца на предмет уровней такролимуса или циклоспорина. Transplant Proc. 2011; 43: 2241–3.
CAS
Статья
Google ученый
Джуньент М., Такер К.Л., Смит К.Э., Гарсия-Риос А., Маттеи Дж., Лай К.К., Парнелл Л.Д., Ордовас Дж. М.. Влияние полиморфизма ABCG5 / G8 на концентрацию холестерина ЛПВП в плазме зависит от привычки курить, согласно Бостонскому исследованию здоровья в Пуэрто-Рико. J Lipid Res. 2009. 50: 565–73.
CAS
Статья
Google ученый
Коловоу В., Марваки А., Каракоста А., Василопулос Г., Калогиани А., Маврогени С., Дегианнис Д., Марваки С., Коловоу Г. Ассоциация пола, полиморфизмов гена ABCA1 и профиля липидов у молодых медсестер Греции.Lipids Health Dis. 2012; 11: 62.
CAS
Статья
Google ученый
Li Q, Yin RX, Wei XL, Yan TT, Aung LH, Wu DF, Wu JZ, Lin WX, Liu CW, Pan SL. АТФ-связывающий кассетный транспортер полиморфизмы G5 и G8 и несколько факторов окружающей среды с уровнями липидов в сыворотке. PLoS One. 2012; 7: e37972.
CAS
Статья
Google ученый
Ма XY, Liu JP, Song ZY.Связь полиморфизма АТФ-связывающего кассетного транспортера A1 R219K с уровнем ХС-ЛПВП и риском ишемической болезни сердца: метаанализ. Атеросклероз. 2011; 215: 428–34.
CAS
Статья
Google ученый
Miura K, Yoshiura K, Miura S, Shimada T, Yamasaki K, Yoshida A, Nakayama D, Shibata Y, Niikawa N, Masuzaki H. Сильная связь между ушной серой человека и секрецией апокринного молозива из молочная железа.Hum Genet. 2007; 121: 631–3.
Артикул
Google ученый
Накано М., Мива Н., Хирано А., Йошиура К., Ниикава Н. Сильная связь подмышечного осмидроза с влажным типом ушной серы, определяемая генотипированием гена ABCC11. BMC Genet. 2009; 10: 42.
Артикул
Google ученый
Oh IH, Oh C, Yoon TY, Choi JM, Kim SK, Park HJ, Eun YG, Chung DH, Kwon KH, Choe BK.Ассоциация полиморфизмов гена CFTR с папиллярным раком щитовидной железы. Oncol Lett. 2012; 3: 455–61.
CAS
Статья
Google ученый
Ота М., Фуджи Т., Немото К., Тацуми М., Моригути Ю., Хашимото Р., Сато Н., Ивата Н., Кунуги Х. Полиморфизм гена ABCA1 придает предрасположенность к шизофрении и связанным с ней изменениям мозга. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011; 35: 1877–83.
CAS
Статья
Google ученый
Сайнц Дж., Рудольф А., Хайн Р., Хоффмайстер М., Бух С., фон Шонфельс В., Хампе Дж., Шафмайер С., Фольцке Х., Франк Б. и др. Ассоциация генетических полиморфизмов генов ESR2, HSD17B1, ABCB1 и SHBG с риском колоректального рака. Endocr Relat Cancer. 2011; 18: 265–76.
CAS
Статья
Google ученый
Sundar PD, Feingold E, Minster RL, DeKosky ST, Kamboh MI. Гендерно-специфическая ассоциация полиморфизмов переносчика 1 АТФ-кассеты (ABCA1) с риском позднего начала болезни Альцгеймера.Neurobiol Aging. 2007. 28: 856–62.
CAS
Статья
Google ученый
Йошиура К., Киношита А., Исида Т., Ниноката А., Исикава Т., Канаме Т., Баннаи М., Токунага К., Сонода С., Комаки Р. и др. SNP в гене ABCC11 является детерминантой типа ушной серы человека. Нат Жене. 2006; 38: 324–30.
CAS
Статья
Google ученый
Montgomery SB, Goode DL, Kvikstad E, Albers CA, Zhang ZD, Mu XJ, Ananda G, Howie B, Karczewski KJ, Smith KS, et al.Происхождение, эволюция и функциональное влияние вариантов с короткой инсерцией-делецией, идентифицированных в 179 геномах человека. Genome Res. 2013; 23: 749–61.
CAS
Статья
Google ученый
Levenstien MA, Klein RJ. Прогнозирование функционально важных классов SNP на основе отрицательного отбора. BMC Bioinformatics. 2011; 12:26.
Артикул
Google ученый
Saunders MA, Liang H, Li WH.Полиморфизм человека в сайтах-мишенях микроРНК и микроРНК. Proc Natl Acad Sci U S. A. 2007; 104: 3300–5.
CAS
Статья
Google ученый
Hsiao YH, Bahn JH, Lin X, Chan TM, Wang R, Xiao X. Альтернативный сплайсинг, модулируемый генетическими вариантами, демонстрирует ускоренную эволюцию, регулируемую высококонсервативными белками. Genome Res. 2016; 26: 440–50.
CAS
Статья
Google ученый
Троусдейл Дж. MHC, болезнь и отбор. Immunol Lett. 2011; 137: 1–8.
CAS
Статья
Google ученый
Suo C, Xu H, Khor CC, Ong RT, Sim X, Chen J, Tay WT, Sim KS, Zeng YX, Zhang X и др. Естественный положительный отбор и генетическое разнообразие север-юг в Восточной Азии. Eur J Hum Genet. 2012; 20: 102–10.
CAS
Статья
Google ученый
Jiang Y, Zhang H. Непараметрический тест, основанный на оценке склонности, выявляющий генетические варианты, лежащие в основе биполярного расстройства. Genet Epidemiol. 2011; 35: 125–32.
Артикул
Google ученый
Росс К.А. Доказательства конверсии и делеции соматических генов при биполярном расстройстве, болезни Крона, ишемической болезни сердца, гипертонии, ревматоидном артрите, диабете 1 типа и диабете 2 типа. BMC Med. 2011; 9: 12.
Артикул
Google ученый
Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. Интегрированная карта генетических вариаций из 1092 геномов человека. Природа. 2012; 491: 56–65.
Артикул
Google ученый
Zhou Y, Ingelman-Sundberg M, Lauschke VM. Распространение аллелей цитохрома P450 по всему миру: метаанализ проектов секвенирования в масштабе популяции. Clin Pharmacol Ther. 2017; 102: 688–700.
CAS
Статья
Google ученый
Bank PCD, Swen JJ, Guchelaar HJ. Применение фармакогеномики в повседневной клинической практике. Adv Pharmacol. 2018; 83: 219–46.
Артикул
Google ученый
Fujikura K, Ingelman-Sundberg M, Lauschke VM. Генетическая изменчивость супергенного семейства цитохрома P450 человека. Pharmacogenet Genomics. 2015; 25: 584–94.
CAS
Статья
Google ученый
Охаши Дж., Нака И., Цучия Н. Влияние естественного отбора на SNP ABCC11, определяющее тип ушной серы. Mol Biol Evol. 2011; 28: 849–57.
CAS
Статья
Google ученый
Радж Т., Кучру М., Реплогл Дж. М., Райчаудхури С., Незнакомец Б. Е., Де Джагер ПЛ. Аллели общего риска воспалительных заболеваний являются мишенями недавнего положительного отбора.Am J Hum Genet. 2013; 92: 517–29.
CAS
Статья
Google ученый
Амато Р., Пинелли М., Монтичелли А., Марино Д., Миле Дж., Кокоцца С. Сканирование всего генома для выявления признаков дифференциации человеческой популяции и их взаимосвязи с естественным отбором, функциональными путями и заболеваниями. PLoS One. 2009; 4: e7927.
Артикул
Google ученый
Musumeci L, Arthur JW, Cheung FS, Hoque A, Lippman S, Reichardt JK.Однонуклеотидные различия (SND) в базе данных dbSNP могут привести к ошибкам в исследованиях генотипирования и гаплотипирования. Hum Mutat. 2010; 31: 67–73.
CAS
Статья
Google ученый
Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. Карта вариаций генома человека по результатам популяционного секвенирования. Природа. 2010; 467: 1061–73.
Артикул
Google ученый
Voight BF, Kudaravalli S, Wen X, Pritchard JK. Карта недавнего положительного отбора в геноме человека. PLoS Biol. 2006; 4: e72.
Артикул
Google ученый
Хуанг Д. В., Шерман Б.Т., Лемпицки РА. Систематический и комплексный анализ больших списков генов с использованием ресурсов биоинформатики DAVID. Nat Protoc. 2009. 4: 44–57.
CAS
Статья
Google ученый
Huang d W., Sherman BT, Lempicki RA. Инструменты обогащения биоинформатики: пути к всестороннему функциональному анализу больших списков генов. Nucleic Acids Res. 2009; 37: 1–13.
Артикул
Google ученый
Полиморфизмы, эпигенетика и что-то между ними
В самом широком смысле сказать, что фенотип является эпигенетическим, предполагает, что он возникает без изменений в последовательности ДНК, но передается по наследству через деление клеток и иногда от одного поколения организма к другому. .Поскольку регуляторные изменения генов часто происходят в ответ на стимулы окружающей среды и могут сохраняться в дочерних клетках, растет ожидание того, что чей-то опыт может иметь последствия для последующих поколений и, таким образом, повлиять на эволюцию, отделяя выбираемый фенотип от лежащего в его основе наследственного генотипа. Но риск такого чрезмерного использования «эпигенетики» — это сочетание реальных случаев наследственной генетической информации, не связанной с последовательностями, с тривиальными способами регуляции генов. Взгляд на термин «эпигенетический» и некоторые проблемы, связанные с его растущей распространенностью, свидетельствует в пользу более сдержанного и точного набора определяющих характеристик.Кроме того, вопросы, возникающие о том, как мы определяем аспект эпигенетического наследования «независимость от последовательностей», предполагают форму эволюции генома в результате индуцированных полиморфизмов в повторяющихся локусах (например, рДНК или гетерохроматин).
1. Эпигенетика и эволюция
Важность полиморфизмов последовательностей в эволюции фундаментальна и неопровержима. Роль эпигенетической регуляции генов изучена значительно хуже. С этой точки зрения я не буду пытаться суммировать все исследования, которые внесли свой вклад в наше нынешнее понимание эпигенетики; вместо этого я объединю несколько важных исследований, взятых в частности, но не исключительно из исследований дрозофилы, чтобы осветить, как общая и последующая «эпигенетическая» регуляция генов может быть результатом индуцированного полиморфизма.Включение индуцированного полиморфизма в арсенал эпигенетических механизмов регуляции генов может заставить нас пересмотреть наши определения, но согласуется с текущим и историческим использованием «эпигенетики» и может предоставить новый механизм для понимания того, насколько стабильными могут быть изменения в экспрессии генов. установлено и поддерживается.
Чтобы понять роль эпигенетики в эволюции, необходимо рассмотреть определения как эволюции, так и эпигенетики. Для целей любого обсуждения, связывающего эти два, «эволюция» должна расширяться, чтобы включать изменение частоты фенотипических вариантов независимо от лежащих в основе аллельных вариантов.Это небольшое отклонение от ориентированного на последовательность взгляда на изменения частот аллелей в развивающихся популяциях, но, по иронии судьбы, больше соответствует первоначальному использованию термина «эпигенетический» для описания абстрактных процессов, которые производят фенотип из генотипа до выяснения причин. центральная догма, регуляция генов и генетика развития. Итак, «эпигенетика» — это примеры изменений в регуляции генов, которые не соответствуют лежащим в основе изменениям в нуклеотидной последовательности. Стоит остановиться на том, что подразумевается под «изменениями в нуклеотидной последовательности», что я и сделаю позже.В общем, изменения в нуклеотидной последовательности являются «полиморфизмами», хотя обычно их называют «генетическими», чтобы противопоставить их «эпигенетическим». Однако это неправильное использование, а генетика — это изучение наследования и изменчивости, независимо от их причины; полиморфизм и эпигенетика — это подмножества генетики, и, как я надеюсь убедить вас, они не являются ни исключительными, ни исчерпывающими (рис. 1).
Понимание совместного вклада в эволюцию полиморфизма и эпигенетики, особенно последней, требует понимания разницы между ними.Это различие является глубоким, поскольку, хотя считается, что полиморфизмы характеризуются случайными, постоянными и хорошо изученными изменениями генетической информации, эпигенетическая регуляция генов более изменчива и, следовательно, стала включать индуцированные и обратимые изменения наследственных признаков. Это порождает популярное (но несправедливое) мнение Жана-Батиста де Ламарка о том, что наши собственные действия или опыт могут повлиять на наше потомство. Ламарк предполагал, что организм эволюционирует, передавая свой опыт потомству.На первый взгляд, наследование приобретенных характеристик соответствовало медленной смене видов с течением времени. Лишь после того, как Вейсманн сформулировал трудности, связанные с шеей жирафа, обсуждая свой опыт со спермой жирафа, ламарковский механизм эволюции был отброшен. Возрождение ламарковских моделей эволюции произошло недавно по ряду причин. Во-первых, есть четкие примеры наследования информации вне последовательности ДНК, что открыло возможность влияния опыта, влияющего на экспрессию генов, и передачи таких изменений в экспрессии потомству.Во-вторых, эта гипотетическая модель не только возможна, но и является еретической, провокационной и, следовательно, захватывающей. В-третьих, возможно, многие из нас чувствуют себя более чем немного виноватыми, высмеивая в остальном выдающегося ученого, который ошибся.
2. Что такое эпигенетика?
Четкое, краткое и исчерпывающее определение эпигенетики сложно сформулировать не потому, что это сложно per se , а потому, что этот термин расширился за последнее десятилетие и начал включать в себя вещи, которые, возможно, не являются эпигенетическими. .Чтобы прояснить ситуацию, Янгсон и Уайтлоу дали убедительное описание разницы между двумя типами «эпигенетики»: трансмиссивными изменениями экспрессии (которые они назвали «трансгенерационными эпигенетическими эффектами») и трансмиссивными модификациями хромосом («трансгенеративным эпигенетическим наследованием») [1 , 2]. Они пытались разделить два очень разных набора явлений, которые описываются как эпигенетические. Многие случаи «эпигенетики» в недавней литературе попадают в первую категорию и вообще не являются эпигенетическими, а скорее являются примерами регуляции генов половых клеток.Чтобы быть значимым отличием от простых форм регуляции генов «экспрессия фактора транскрипции и усиления усилителя», эпигенетические явления должны проявлять три характеристики: они должны проявляться как (1) наследственные генетические изменения, которые (2) связаны с хромосомами, но (3) не основаны на последовательности ДНК. . Это критерии, от которых не следует отказываться, но их следует оценить.
Вторая характеристика важна, потому что она составляет сущность эпигенетического наследования. Почему? Потому что, если бы эпигенетика не требовала ассоциации хромосом, каждый генетический путь, включающий петлю положительной обратной связи, был бы эпигенетическим.Специфический для самок летальный сплайсинг у Drosophila с образованием более активного сексуально-летального фактора сплайсинга будет считаться эпигенетическим. Бактериальная экспрессия LacY, пермеазы лактозы, повышающая чувствительность к дальнейшему воздействию лактозы, будет считаться эпигенетической. Аутофосфорилирование CaMKII при наблюдении всплеска кальция можно считать эпигенетическим. Вызванная супрессором безволосая экспрессия Notch, активатора супрессора-безволосого, будет считаться эпигенетической. Короче говоря, почти каждую генетическую сеть можно считать эпигенетической, и «эпигенетика» не будет существенно отличаться от «генной регуляции».«Поскольку белки, липиды, РНК, промежуточные продукты метаболизма и даже токсины проходят через клеточное деление в цитоплазме, тривиально говорить, что их эффекты« наследуются », и ошибочно делать вывод, что клетки сохраняют последствия своих предшественников». переживания обязательно эпигенетичны.
Не требуя хромосомного наследования эпигенетических явлений, экспрессии в зародышевой линии было бы достаточно для разграничения любого генетического пути как эпигенетического, что служило бы просто для переименования тех генов, которые экспрессируются во время создания яйцеклеток и сперматозоидов.Неудивительно, что такие сети могут охватывать несколько поколений организмов; в конце концов, именно генетика (и опыт) матери создают яйцеклетку, а генетика (и опыт) отца создают сперму; изменение этих процессов, безусловно, приведет к изменениям в следующем поколении. Биология млекопитающих еще больше усугубляет проблему, поскольку в поздних сроках беременности могут участвовать три концентрических организма: к концу беременности самки млекопитающих содержат половину генома всех своих потенциальных внуков; Ооциты, содержащие эти пронуклеусы, заполнены генными продуктами, созданными их матерями из питательной среды, предоставленной их бабушкой.Многие случаи, называемые эпигенетическими, вместо этого представляют собой эту форму регуляции трансгенеративного гена.
Чтобы отличить широкие концепции «памяти» или «потенцирования» в регуляции генов от специфического эпигенетического наследования, необходимо показать, что эпигенетические факторы, изменяющие активность гена, специфически картируются в регулируемом хромосомном локусе. Экспериментально демонстрируется регуляция эпигенетического гена , когда ДНК нарушает закон массового действия : две идентичные последовательности могут действовать по-разному, несмотря на идентичность их последовательностей и белков, которые с ними связываются.Концептуально, если «наивная» ДНК будет введена в систему, будет ли она вести себя так же, как существующие ДНК? Если так, то это не эпигенетика. На практике это легче всего показать, продемонстрировав, что идентичные фрагменты ДНК (гомологи, дупликации, трансгены, особи повторяющихся массивов генов и т. Д.) Обладают разным поведением. Это было показано для идентичности центромер [3, 4], геномного импринтинга у млекопитающих [5], растений [6-8], грибов [9, 10], нематод [11] и насекомых [12-14]; именно это требование не проверяется многими примерами «эпигенетики».Сильная связь между эпигенетикой и структурой хроматина только способствовала объединению этих терминов. Нет ничего необычного в том, чтобы найти термин «эпигенетический» в исследованиях, которые просто показывают изменения в гистоновых модификациях гена, возможно даже ацетилирование, без экспериментов, которые проверяют наследуемость, полиморфизм последовательностей или хромосомную ассоциацию.
3. Существует ли эпигенетика?
Изменения в нуклеотидной последовательности, приводящие к фенотипическим вариантам, ясны, установлены и составляют саму основу неодарвиновского синтеза, который объединил дарвиновские теории вариации и отбора с правилами наследования Менделя.Что было и остается волшебным в эпигенетике, так это то, что можно увидеть существенные вариации без очевидных основных изменений в последовательности нуклеиновых кислот, и поэтому такие изменения относительно нестабильны. Первое, что привлекло внимание к эпигенетическому наследованию, так это различное поведение идентичных геномов в пестроте в результате косупрессии, которая инактивирует дублированные копии генов в растениях, эффекты положения дрозофилы, вызванные гетерохроматином [15], или соматический мозаицизм из-за инактивации Х-хромосомы. у самок млекопитающих.Эти различия в фенотипах не были бы удивительными, если бы они были обусловлены различиями в последовательности ДНК.
Но насколько тщательно мы проверяли идентичность последовательностей в этих случаях? Представьте себе гипотетическую ситуацию. Что, если для создания центромеры требуется фермент (центромера?), Чтобы разрезать ДНК и вставить определенную последовательность, необходимую и достаточную для установления активности центромеры? Что, если бы неоцентромеры были случаями редкой случайной экспрессии и активности центромеразы? Что, если потеря центромерной активности в дицентрических Робертсоновских хромосомах слияния была доказательством обратимости центромеразы? Гипотетическое существование центромеразы, конечно, не нужно, учитывая то, что мы знаем о центромерных гистонах и структуре хроматина, но показательно, что во многих случаях специфический индуцированный полиморфизм даже не рассматривается.Мы придерживаемся мнения, что случайная мутация — единственный механизм, позволяющий изменять последовательность ДНК, и поэтому быстрые, индуцированные и обратимые изменения поведения хромосом должны происходить без изменения последовательности. Но это предполагает более четкие линии в определении «последовательности», чем существуют на самом деле, и игнорирует многие хорошо установленные наблюдения.
Рассмотрим переключение типа спаривания у Schizosaccharomyces pombe. Переключение происходит, когда тихая кассета информации из локуса «хранения» передается в активный локус типа спаривания [16–18].Механизм переключения требует метки, вероятно, разрыва или рибонуклеотида на одной цепи [19]. Прослеживание происхождения этой цепи показало, что измененная цепь происходит из переключенного локуса в предыдущем поколении. В результате переключение ограничено по частоте и направлению. Рибонуклеотид в цепи дезоксирибонуклеиновой кислоты действительно является удивительным способом передачи информации о хромосоме, но, тем не менее, он является генетическим: он наследуется и является следствием. И что самое удивительное, это индуцируется.
Рассмотрим также геномный импринтинг у млекопитающих. Действительно ли геномный импринтинг эпигенетичен? Хотя это, пожалуй, наиболее приемлемая форма эпигенетики, можно утверждать, что это не так, по тривиальным номенклатурным причинам: считаете ли вы 5-метилцитозин как цитозин или как пятое основание, которое просто требует дополнительного включения (связанное с репликацией ДНК-метилтрансфераза)? Хотя ваш ответ может кое-что рассказать о вашей философии, он влияет на то, как мы думаем об эпигенетических механизмах.Если считать 5-метилцитозин пятым основанием, то материнские и отцовские аллели геномно импринтированных генов действительно полиморфны. Можем ли мы также считать полиморфизмом дегидроксилирование или дегликозилирование? Рассмотрение этих случаев индуцированного полиморфизма исключило бы как переключение типа спаривания S. pombe, так и импринтинг в локусе Medea (где метилирование цитозина вызывает разрыв цепи на одном гомологе, облегчая его от сайленсинга) как эпигенетические. И почему бы нет? Селеноцистеин — это аминокислота, хотя рибосома требует обширной сложной системы для ее включения [20, 21].Метилцитозин химически и генетически отличается от цитозина; для его включения требуется лишь обширная и продуманная система. Разорванная цепь ДНК снова химически и генетически отлична. Это забавный аргумент, но он кажется чрезмерно надуманным и ненужным и, вероятно, немного странным. Дело не в том, что нам нужно удалить эти случаи из списка эпигенетики, а скорее в том, что мы должны учитывать, что мы подразумеваем под «последовательностью», когда используем ее в качестве ключевого критерия, отличающего «эпигенетику» от «полиморфизмов».«В этой серой зоне много ландшафта.
4. Что-то среднее между
Понимание того, как и почему мы определяем «последовательность» и «эпигенетичность», важно при классификации способов регуляции генов. Но такие соображения также раскрывают понимание того, как эти явления могут взаимодействовать, и заставляют нас задуматься о том, насколько важным может быть индуцированный полиморфизм в эволюции. Вышеприведенные примеры — Medea, тип спаривания и импринтинг — все это случаи индуцированного полиморфизма, который приводит к изменениям генетической активности последовательности.Тот факт, что они «независимы от последовательности», является артефактом нашей систематической ошибки ACGT-последовательности. Тем не менее, кажется сомнительным, что эти несколько примеров сами по себе опровергают наши взгляды на эволюцию. Во-первых, такие способы эпигенетической регуляции генов, по-видимому, необычны. Подсчитано, что у млекопитающих есть, возможно, сотни импринтированных локусов, а у растений всего лишь один. Во-вторых, это не случаи наличия / отсутствия генетических путей, а скорее смещения экспрессии различных аллелей, и поэтому сиблинги не отличаются заметно из-за этого способа регуляции; импринтированные гены по существу гаплоидны и поэтому мало чем отличаются от сцепленных с полом генов с точки зрения эволюции.В-третьих, они сбрасываются через одно поколение. Поэтому трудно представить, что эти формы эпигенетического наследования движут эволюцией глубоким или новым образом.
Существуют ли примеры индуцированного полиморфизма, которые являются широко распространенными, последовательными и долгоживущими и поэтому могут повлиять на эволюцию генома?
Почти половина геномов многих популярных многоклеточных животных высоко полиморфны, но эти полиморфизмы остаются незамеченными при общегеномных ассоциациях, локусах количественных признаков и популяционных генетических исследованиях.Эта часть, гетерохроматин — альфоидные и бета-повторы, мобильные элементы, сателлиты, повторяющаяся последовательность и так далее, обычно связанные с центромерами — не поддаются нашим современным подходам к геномике. Гетерохроматин может содержать сотни, тысячи и даже миллионы копий простых (например, AATAT, AAGAG и AAGAGAG) повторов [22–26]; следовательно, они не могут быть легко клонированы, секвенированы или собраны с использованием методов, направленных на секвенирование всего генома. Фактически, определение «целого» было изменено, чтобы игнорировать эту половину генома [27–29].Количественная оценка числа копий повторов является обременительной и неточной, и наткнуться на редкие полиморфизмы последовательностей в гомогенных блоках сателлитной ДНК — это удача [30]. Поэтому трудно оценить степень различий или скорость полиморфизма в этой значительной части генома.
Гетерохроматин был впервые описан Эмилем Хайцем в 1920-х и 1930-х годах. В то время его отличительной чертой было гетеропикнотическое окрашивание, которое до сих пор, возможно, является лучшим определением.Впоследствии было обнаружено, что гетерохроматин обычно поздно реплицируется, репрессивен для экспрессии генов и обогащен специфическими модификациями ДНК и гистонов, которые его упаковывают, хотя есть исключения для всех этих особенностей [15, 31, 32]. Все согласны с тем, что гетерохроматин легко образуется на сильно повторяющейся последовательности и существует в виде комплекса с белками гетерохроматина (например, гистоновыми метилтрансферазами, HP1 и, возможно, РНК). Генетические и мутационные манипуляции, которые изменяют количество повторяющейся последовательности или белковых компонентов, демонстрируют естественный баланс между последовательностью и белковыми компонентами при формировании гетерохроматина [33–37].Избыточная последовательность ставит под угрозу образование гетерохроматина в другом месте, конкурируя за ограниченные белки гетерохроматина. Увеличение или уменьшение белков гетерохроматина увеличивает или уменьшает легкость образования гетерохроматина или увеличивает или уменьшает количество последовательности, которая может быть упакована как гетерохроматин.
Малик и Хеникофф описали свой взгляд на конкретный пример эволюционного баланса в центромерном хроматине (или «центрохроматине») [38-40]. Они предполагают совместную эволюцию расширения последовательности и связывания ДНК с помощью центромерного гистона Cid.Избыточная центромерная ДНК связывается с Cid, и изменения в связывании (или экспрессии) Cid приводят к изменению центромерной последовательности. Это может быть примером более широкого механизма расширения и сжатия, ограниченного (или обнародованного) характеристиками ДНК-связывающих белков, которые стабилизируют повторяющуюся последовательность. Смесь множественных полиморфных простых повторов в геноме [25, 26, 41, 42] может быть стабилизирована смесью выделенных или перекрывающихся белков гетерохроматина [43–48]. Баланс между последовательностями и белками, которые вместе образуют гетерохроматин, как ожидается, будет важным, потому что белковые компоненты гетерохроматина играют двойную роль в качестве общих регуляторов транскрипции [49, 50].Гены переключаются между «гетерохроматиноподобными» и «эухроматиноподобными», когда они переключаются между молчанием и экспрессией во время развития или в ответ на стимулы окружающей среды. Мутации в генах, кодирующих эти белковые компоненты, часто действуют доминантно, что позволяет предположить, что их доза имеет значение [34, 36]. Легко представить себе трехсторонний баланс между гетерохроматиновой последовательностью, гетерохроматиновыми белками и механизмами регуляции эухроматических генов. Это предсказывает, что полиморфизм числа копий гетерохроматин-образующей последовательности может влиять на регуляцию генов по всему геному.
Было очень сложно проверить, являются ли полиморфизмы числа копий последовательными, потому что существует несколько молекулярно-генетических инструментов, которые позволяют манипулировать числом копий в других изогенных фонах. Из классических исследований на Drosophila, где ДНК и белковые компоненты гетерохроматина легко манипулировать, мы знаем, что количество гетерохроматиновой последовательности в клетке резко влияет на сенсибилизированные гены вариегатности [33, 51, 52]. В крайнем случае, несколько лишних гетерохроматиновых хромосом смертельны [53].Хотя причина остается неясной, можно представить себе такое нарушение баланса между последовательностями и белками, которое вызовет массовую неправильную регуляцию многих генов. Y-хромосомы, захваченные из диких популяций, различаются по своей способности влиять на вызванное гетерохроматином изменение положения и экспрессию эухроматических генов в другом месте генома [54–56], но при этом содержат очень мало генов, кодирующих белок [57–59], что убедительно свидетельствует о том, что гетерохроматин полиморфизмы, возможно, полиморфизмы числа копий, влияют на экспрессию генов по всему геному.Наша работа вызвала изменение количества копий в одном повторе, рибосомной ДНК (рДНК) [60]. РДНК имеет прецедент для фенотипической изменчивости растений, вызванной помещением [61, 62], но, не будучи способной вызвать изменения в рДНК, было трудно проверить этот феномен в дальнейшем. У мух, однако, индуцированная вариация числа копий имеет последствия для индуцированного гетерохроматином вариэгации эффекта положения и экспрессии генов по всему геному [63, 64]. Эти вариации в регуляции генов перекрываются с вариациями, наблюдаемыми в изолированных природных Y-хромосомах [54, 64], предполагая, что значительная часть естественной вариации числа копий повторов рДНК [65, 66] может вносить вклад в фенотипическую вариативность в естественных популяциях.Не менее важно, что большая часть дисперсии, которая отображается на Y-хромосоме, не отображается на рДНК, предполагая, что большая часть фенотипической дисперсии отображается на другие последовательности на Y, возможно, на другие повторы, которые менее экспериментально поддаются манипулированию.
Естественные вариации числа копий повторяющейся последовательности могут играть роль в эволюции, но уникальная динамическая биология рДНК предполагает более захватывающую возможность. Изменения в количестве копий могут быть вызваны и унаследованы.
РДНК содержит перемежающиеся активные и неактивные гены рРНК и, таким образом, содержит характеристики как эухроматина, так и гетерохроматина в некоторых клетках.Физическим проявлением огромной экспрессии и процессинга рРНК является ядрышко. Стабильность этих длинных участков прямых повторов в ядре, вероятно, обусловлена гетерохроматической упаковкой подмножества повторов. Пенг и Карпен наблюдали множественные ядрышки в постмитотических клетках животных, несущих мутации генов супрессоров пестроты, которые кодируют белковые компоненты гетерохроматина и регулируют рДНК [67]. Их результаты предполагают, что повторяющаяся последовательность, не упакованная как гетерохроматин, нестабильна и склонна к повреждению / репарации или внутрихромосомной рекомбинации.Наши эксперименты показали, что мутация генов-супрессоров пестролистности приводит к дестабилизации и снижению числа копий рДНК через митоз [63]. Мы далее количественно оценили потерю в соме, а также показали, что потеря наблюдалась через зародышевую линию, что привело к постоянному снижению количества копий рДНК в популяции после воздействия мутации, которая нарушает образование гетерохроматина. Недавно мы показали, что мутация репрессора экспрессии рДНК (CCCTC-Factor, или CTCF) также дестабилизирует рДНК, приводя к необратимой потере [68].Эти результаты согласуются с гетерохроматиноподобным замалчиванием, стабилизирующим повторяющуюся последовательность ДНК, и балансом между повторяющимися последовательностями и белковыми компонентами, которые их регулируют.
У дрозофилы рибосомная ДНК является неотъемлемой частью, потому что ее динамизм не имеет себе равных. Это наиболее экспрессируемый набор генов в геноме [69], координирует активность всех трех полимераз, естественным образом сокращается за счет образования внехромосомных кругов [70], может восстанавливаться посредством мейотического увеличения или соматического псевдомагнетизма [71–75]. , и может компенсировать свой выход за счет изменения скорости удлинения и, возможно, скорости инициирования [76, 77].Он обладает соседними копиями, которые обладают гетерохроматической и эухроматической структурами хроматина [76, 78–80]. Как центральный орган в способности к синтезу белка, он также реагирует на статус питания, чувствителен к токсинам и лекарствам и подвержен нестабильности из-за изменений продуктов генов, необходимых для его регуляции [81–87]. Изменение регуляции рДНК посредством мутации или лечения лекарствами влияет не только на выход рРНК, но и на стабильность [88–90]. Следовательно, изменение активности белкового компонента гетерохроматина может повлиять на количество копий последовательности, с которой он связывается.
Динамизм (рДНК) и баланс (гетерохроматиновой последовательности и белков) создают ситуацию гомеостаза гетерохроматина (рис. 2). Последовательности защищены от потери за счет упаковки в виде гетерохроматина. Потеря белка (или снижение активности белка, стрелка «а») дестабилизирует повторяющуюся ДНК (белое состояние) и приведет к потере, восстанавливая равновесие (стрелка «b» в серое состояние). Точно так же избыточная последовательность будет возвращаться через потерю, если не будет достаточного количества белка, чтобы упаковать ее для стабильности.Но избыток белка имеет последствия, поскольку любой гетерохроматиновый белок, не связанный в конститутивной последовательности, будет изменять экспрессию гена по всему геному (темно-серое состояние), благоприятствуя либо снижению экспрессии / активности белка (стрелка «c»), либо увеличению повторяющейся последовательности (стрелка «Г») для восстановления баланса (светло-серые состояния). В целом, нестабильность повторяющейся последовательности и следствие избытка гетерохроматиновых белков создает множественные состояния, которые уравновешивают факторы и естественным образом приводят к уравновешиванию количества повторяющихся последовательностей и экспрессии белка.Конечно, можно ожидать, что любые внешние факторы, влияющие на активность белка гетерохроматина, приведут к индуцированным и наследственным изменениям в количестве повторяющихся копий ДНК. РДНК особенно чувствительна к индуцированному полиморфизму числа копий, поскольку на него влияет статус питания на протяжении всей жизни организма, а число копий рДНК превышает то, что требуется для трансляции, что позволяет некоторую пластичность числа копий без чрезмерных недостатков.
На первый взгляд, индуцированный полиморфизм числа копий похож на эпигенетическую модификацию (особенно если невозможно легко секвенировать и собрать повторяющиеся ДНК), а способность повторяющихся последовательностей изменять число копий относительно легко добавляет степень летучести, характерную для эпигенетическая регуляция генов.В отличие от многих форм трансгенерационных эффектов регуляции генов, индуцированные полиморфизмы числа копий связаны с хромосомами и, таким образом, являются как наследуемыми, так и селектируемыми. В отличие от эпигенетической регуляции импринтированных или инактивированных хромосом, индуцированный полиморфизм числа копий может быть унаследован от нескольких поколений. Но, как и трансгенерационные и эпигенетические эффекты, роль индуцированного полиморфизма только начинает рассматриваться в эволюции. Такое исследование, вероятно, будет выполнено на простых организмах, таких как Drosophila, которые имеют относительно простую архитектуру рДНК [91, 92].Напротив, у людей есть несколько массивов рДНК, которые часто меняются в размере [93], и сложная регуляция, которая делает одни массивы активными, а другие неактивными, означает, что может пройти некоторое время, прежде чем мы поймем, как полиморфизмы рДНК и нестабильность рДНК [94] вносят вклад в фенотип. различия в человеческой популяции или этиологии болезни.
5. Является ли рДНК особенной?
Индуцированный полиморфизм числа копий рДНК предлагает удобный механизм, с помощью которого изменения могут быть унаследованы, хотя здесь применимы те же возражения, что и в отношении вызванных окружающей средой изменений в экспрессии генов, которые вытягивают шею Ламарка: как это влияет на зародышевую линию? В случае индуцированного полиморфизма половые клетки могут быть более, а не менее чувствительны к индуцированным изменениям в составе гетерохроматина по трем причинам.Во-первых, во многих случаях экспрессия генов в этих типах клеток ограничена. Устойчивость белков гетерохроматина или наличие достаточного количества генного продукта, способного выдержать колебания активности гена, может быть меньше в этих типах клеток. Во-вторых, по крайней мере, у мужчин геном лишен большинства компонентов соматического хроматина в пользу упаковки белков и полиаминов. Это может повысить чувствительность таких хромосом к перестройкам ДНК или специфически пометить некоторые области для гипервариабельности. В-третьих, половые клетки естественным образом подвергаются рекомбинации с высокой скоростью.Хорошо известно, что изменения в микросателлите и количестве копий рДНК происходят в мейозе, в то время как одни и те же последовательности относительно стабильны в митозе. Задача состоит в том, чтобы понять, что идентифицирует ген как «чувствительный к количеству копий рДНК», потому что это будут те гены, которые выбраны для фенотипической изменчивости в ответ на изменение количества копий рДНК.
Мы еще не понимаем, отличаются ли повторяющиеся последовательности от «неэкспрессированных» последовательностей способностью вызывать изменения, но мы знаем из мутационного и молекулярного анализов, что «гетерохроматин» не является монолитным и более точно рассматривается как множественный ». цвета »[95].Мутации могут влиять на один цвет гетерохроматина, но не на другой [96]. Пять пронумерованных цветностей значительно расширяют наше понимание структуры хроматина, но даже эти пять, вероятно, все еще являются упрощением, вызванным нашей неспособностью разрешить более тонкие различия. Громоздкая работа обнаружила изменения числа копий повторяющейся последовательности в нескольких исследованиях, предполагая, что это может быть очень широко распространенной формой генетической изменчивости [66, 97, 98]. Peng и Karpen показали увеличение очагов репарации повреждений ДНК в гетерохроматине мутантов супрессоров пестролистности в диплоидных клетках [99, 100].Они не идентифицировали репарируемые последовательности, но количество и распределение очагов репарации в ядрах указывало на то, что они не были сгруппированы (т.е. ограничены массивами рДНК). Это наблюдение предполагает, что гетерохроматин, образованный на простых повторах (а не только на высокоэкспрессируемой рДНК), также стабилизируется за счет упаковки в виде гетерохроматина. По мере того, как мы понимаем, что такое гетерохроматин, и по мере того, как становятся доступными инструменты для его более хирургического исследования, мы можем начать распутывать сложные взаимодействия между типами гетерохроматина, когда они изо всех сил пытаются держать друг друга под контролем или объединяться, чтобы отбиваться от общих врагов.
Термин «эпигенетика» может сохранять свои строгие определения связанной с хромосомами генетической информации, не основанной на последовательности, или он может быть расширен за счет включения индуцированных мутаций или генных регуляторных сетей, которые влияют на последующие поколения. В конце концов, все формы регуляции являются генетическими и поэтому важны для понимания того, как сложные, плейотропные и эпистатические генетические взаимодействия способствуют созданию фенотипов. Каким бы ни было определение эпигенетики, наследие состоит в том, что мы не можем понять всеобъемлющий синтез сил, управляющих эволюцией генома, без понимания того, как регулируются все аллели в этом геноме.
Границы | Изучение влияния однонуклеотидных полиморфизмов на трансляцию
Вариация последовательности и экспрессия гена
За последние два десятилетия достижения в области секвенирования генома предоставили беспрецедентный доступ к ландшафту генома человека и позволили документировать вариации последовательностей среди людей. Люди разделяют 99,5% идентичности на уровне геномной последовательности, что означает, что результирующее фенотипическое разнообразие проистекает из оставшейся разницы в 0,5%, а также эпигенетических модификаций.Различия в последовательностях возникают из-за присутствия тандемных повторов с коротким и переменным числом, инсерционных или делеционных полиморфизмов и однонуклеотидных полиморфизмов (SNP) (Mccarroll et al., 2006; Orr and Chanock, 2008). Среди SNP переходы (A ↔ G или C ↔ T) более распространены, чем трансверсии (A ↔ C или T; и G ↔ C или T). В геноме имеется не менее 10 миллионов SNP, встречающихся примерно через каждые 100–300 пар оснований и с частотой аллелей более 1%, что делает их, безусловно, наиболее распространенным типом вариантов в геноме человека (Risch, 2000; Lander et al. ., 2001; Орр и Чанок, 2008). В последнее время наблюдается расцвет исследований общегеномных ассоциаций (GWAS), в которых распространенность конкретных SNP связана с фенотипами или заболеванием (Srinivasan et al., 2016). Кроме того, Атлас ракового генома (TCGA) выявил вариации последовательности между опухолевыми и нормальными клетками, и текущая проблема заключается в различении тех мутаций, которые оказывают влияние на экспрессию генов, чтобы управлять эволюцией опухоли, и нерелевантными мутациями-пассажирами.
Мутации могут изменять все стадии экспрессии генов в зависимости от их геномного местоположения.Когда они присутствуют в элементах регуляции транскрипции, они могут влиять на экспрессию мРНК. Возникая в генах, SNP могут влиять на сплайсинг мРНК, ядерно-цитоплазматический экспорт, стабильность и трансляцию. Когда они присутствуют в кодирующей последовательности и приводят к замене аминокислоты (называемой несинонимичным SNP или мутацией), они могут изменять активность белка. Если мутация синонимична (т.е.не изменяет природу аминокислоты), это может повлиять на скорость трансляции или период полужизни мРНК.Если мутация вызывает преждевременный стоп-кодон, это может привести к продукции усеченного белкового продукта или почти нулевого фенотипа из-за нонсенс-опосредованного распада (Mendell and Dietz, 2001; Nicholson et al., 2010). Проект «Энциклопедия элементов ДНК» (ENCODE) направлен на выявление и каталогизацию функциональных элементов в геноме человека и оказался весьма полезным для понимания потенциального воздействия вариаций последовательностей на экспрессию генов (Consortium, 2012). Однако функциональные последствия вариантов последовательности, которые встречаются в 5′-лидере мРНК [i.е. область перед кодоном инициатора основной открытой рамки считывания (ORF)] и 3′-нетранслируемые области (UTR) (т.е. область ниже основного стоп-кодона ORF) не всегда сразу очевидны и часто не охарактеризованы. . Здесь мы предлагаем некоторые мысли о том, как такие варианты могут влиять на эффективность трансляции мРНК. Мы выделяем отдельные этапы трансляции, на которые могут влиять варианты последовательностей, приводя примеры выбора, когда это уместно, и они суммированы в таблице 1.
ТАБЛИЦА 1. Краткое изложение SNP, описанных в этом исследовании.
Обзор eIF4F-зависимого рекрутирования рибосом
Синтез белка млекопитающих преимущественно регулируется на стадии инициации трансляции, причем лимитирующей стадией является рекрутирование рибосом в мРНК (Figure 1A; Sonenberg and Hinnebusch, 2009). Ключевым посредником на этом этапе является комплекс eIF4F. Субъединица eIF4E связывается со структурой кэпа мРНК, присутствующей на всех цитоплазматических мРНК эукариот. Компонент eIF4G имеет РНК-связывающие домены и стабилизирует взаимодействие eIF4E: cap (Marcotrigiano et al., 2001; Янагия и др., 2009). Структурные элементы РНК разрешаются геликазой РНК-геликазы eIF4A DEAD-box в сочетании с белками, связывающими РНК, eIF4B и / или eIF4H (рис. 1A). Комплекс предварительной инициации 43S (40S рибосома и ассоциированные факторы) (PIC) затем рекрутируется в матрицу мРНК посредством мостиковых взаимодействий между eIF4G и связанным с рибосомами eIF3 (Sonenberg and Hinnebusch, 2009). Этот режим инициации называется cap- или eIF4E-зависимым. Потребность мРНК в eIF4F для рекрутирования рибосом различается и, по-видимому, масштабируется как следствие вторичной структуры 5′-лидера (Pickering and Willis, 2005; Bitterman, Polunovsky, 2015; Hinnebusch et al., 2016). Поскольку считается, что eIF4E является ограничивающим для трансляции, мРНК должны конкурировать за доступ к eIF4F, а мРНК со структурными барьерами в их 5′-лидерной области находятся в невыгодном положении (Pelletier and Sonenberg, 1985; Babendure et al., 2006). Следовательно, изменение ландшафта вторичной структуры в 5′-лидерной области мРНК может существенно влиять на эффективность трансляции, влияя на скорость рекрутирования рибосом (Pelletier et al., 2015). После связывания 43S PIC сканирует 5′-лидерную область мРНК в поисках инициирующего кодона.
РИСУНОК 1. Обзор набора и сканирования рибосом. (A) Cap-зависимая инициация трансляции. Комплекс eIF4F в сочетании с eIF4B и eIF4H служит для подготовки мРНК для привлечения 43S рибосомного комплекса. (B) Влияние uAUG и uORF на сканирование рибосом. При связывании с мРНК 43S PIC (светло-синий) сканирует мРНК в поисках инициатора AUG. Кодон AUG в благоприятном контексте эффективно распознается сканирующей субъединицей 40S, в этот момент субъединица 60S присоединяется и начинается удлинение.Мутации, создающие новые uAUG или uORF, будут влиять на частоту рибосом, которые инициируются в основном кодоне ORF AUG. Положение uORF относительно основного кодона AUG важно для определения основного использования AUG, поскольку расстояние от стоп-кодона uORF и основного AUG определяет время, которое потребуется рибосоме для повторного приобретения eIF2 * GTP. ∗ тройной комплекс Met-тРНК. (C) A G / A SNP в 5′-лидерной области мРНК ERCC5 контролирует экспрессию и ответ на стресс.Аллель A, содержащий мРНК, имеет дополнительную uORF, которая обеспечивает более эффективную трансляцию основной ORF ERCC5 в ситуациях, когда eIF2α фосфорилируется. Подробности см. В тексте.
Второй механизм, с помощью которого 43S PIC может быть рекрутирован в матрицы мРНК, заключается в прямом внутреннем связывании в пределах 5′-лидерной области с внутренним сайтом входа в рибосомы (IRES), устраняя потребность в структуре кэпа. Среди них лучше всего охарактеризованы вирусные IRES, которые были разделены на четыре класса на основе структурного сходства, фактора инициации и / или требований к транс-действующему фактору IRES (Mailliot and Martin, 2018).Некоторые исключительные IRES, такие как вирус паралича сверчка IRES, обходят необходимость в каких-либо факторах инициации и могут напрямую связываться с рибосомой.
Открытие и характеристика IRES в клеточных мРНК представляет значительный интерес, поскольку они участвовали в обеспечении возможности трансляции в условиях, когда кэп-зависимая трансляция нарушена, таких как стресс, апоптоз, ограничение питательных веществ и митоз (Komar and Hatzoglou, 2015). Таким образом, считается, что функция клеточного IRES важна для обеспечения быстрой адаптации к быстро меняющейся среде, что приводит к избирательным трансляционным эффектам.Влияние SNP на клеточную активность IRES может повлиять на реакцию на стресс, такой как гипоксия, тепловой шок, токсины или лекарства (химиотерапия). Кроме того, мутации в клеточных IRES могут приводить к аберрантным трансляционным ответам, вызывающим ряд патологических расстройств, начиная от аутоиммунных заболеваний, нейродегенерации и рака (Holcik and Sonenberg, 2005).
Изменения во вторичной структуре, влияющие на трансляционный вывод
Препятствуя взаимодействиям eIF4F-cap или сканированию рибосом, структурные особенности (например,g., стебель-петли, комплексы РНК-белок, G-квадруплексы) могут действовать как барьеры для инициации трансляции и отрицательно влиять на эффективность трансляции (Pelletier and Sonenberg, 1985; Babendure et al., 2006). Исследование Shen et al. (1999) был одним из первых, кто задокументировал обширное влияние, которое SNP могут оказывать на вторичную структуру мРНК (Таблица 1). Анализ двух SNP в кодирующих областях мРНК, кодирующих аланил-тРНК-синтетазу и репликационный белок А, выявил аллель-специфические структурные особенности, которые влияют на доступность последовательностей.Доказательства того, что такие изменения могут влиять на трансляционный выход, были предоставлены исследованием, оценивающим влияние структур G-квадруплексов, присутствующих в 5′-лидерных регионах, на трансляцию (Beaudoin and Perreault, 2010). SNP (изменение G на C) был идентифицирован в положении 7 G-квадруплекса (критическая область для образования G-квадруплекса) в 5′-лидере мРНК AASDHPPT (аминоадипат-полуальдегиддегидрогеназа-фосфопантетеинилтрансфераза) (Beaudoin и Perreault, 2010). Биофизические эксперименты показали, что образование G-квадруплексов было нарушено, и это было связано с 1.5-кратное увеличение уровня белка в клетках без влияния на уровень мРНК (Beaudoin and Perreault, 2010). Эти эксперименты показывают, что точечные мутации в 5′-лидерных областях, которые изменяют вторичную структуру, могут влиять на трансляцию.
Вторичная структура, расположенная непосредственно после AUG, также может влиять на трансляционный вывод. Например, синонимичный SNP от A до G в кодоне Leu , присутствующий в кодирующей области мРНК катехол- O -метилтрансферазы ( COMT ), был идентифицирован у субъектов с высокой болевой чувствительностью и с повышенным риском развивающееся заболевание височно-нижнечелюстного сустава (Дьяченко и др., 2005; Nackley et al., 2006). Белок COMT отвечает за деградацию катехоламинов и является регулятором восприятия боли. У человека три основных гаплотипа образованы четырьмя SNP в локусе COMT : один расположен в промоторе, а три — в кодирующей области [два синонимичных по his 62 his (C / T) и leu 136 лей, (C / G) и один несинонимичный val 158 met (A / G)] (Nackley et al., 2006). Сообщалось, что основные гаплотипы COMT различались в отношении локальной вторичной структуры мРНК с наиболее стабильной структурой, связанной с самыми низкими уровнями продукции белка (Nackley et al., 2006). Сайт-направленный мутагенез, нарушающий структурный элемент, вызывает увеличение продукции белка. Однако авторы не оценивали напрямую влияние различных последовательностей гаплотипов на скорость трансляции мРНК.
Напротив, вторичная структура может также действовать для увеличения узнавания стартовых кодонов, когда она соответствующим образом расположена ниже инициирующих кодонов — эффект, предположительно, из-за замедления сканирования рибосом и увеличения времени выборки кодонов (Kozak, 1991). Следовательно, изменения последовательности, которые увеличивают образование структуры в проксимальной области нижестоящего AUG, могут увеличивать степень использования AUG.
Вариация 5′-лидерной последовательности и выбор стартовой площадки
SNP влияют на распознавание стартового кодона
Механизм, с помощью которого PIC 43S обнаруживает инициирующий кодон, влияет на то, как SNP, которые генерируют новые или удаляют существующие стартовые кодоны, влияют на инициацию трансляции. Контекст последовательности инициирующего кодона диктует эффективность, с которой он распознается при сканировании 43S PIC (рисунок 1B). Оптимальным контекстом является A / GxxAUGg, называемая консенсусной последовательностью Козака, при этом пурин -3 (относительно A AUG) является наиболее важным детерминантом (Kozak, 1986, 1987a).Предполагается, что мутации, изменяющие этот контекст, повлияют на эффективность выбора стартовой площадки.
Существует множество примеров мутаций, которые изменяют контекст последовательности AUG, чтобы влиять на эффективность трансляции. Одним из таких примеров является описание мутации в гене BRCA1 у 35-летнего пациента, превращающей G в C в положении -3 относительно инициирующего кодона BRCA1 AUG (Signori et al., 2001). Эта мутация изменяет оптимальный пурин на менее подходящий пиримидин и связана со спорадическим раком груди и яичников (Hall et al., 1990; Сабо и Кинг, 1995). In vitro и in vivo Исследования экспрессии репортерных мРНК, несущих аллель C, показали снижение экспрессии белка на 30–50% по сравнению с контрольными мРНК, несущими аллель G. Кроме того, транскрипт, несущий аллель G, был связан с более тяжелыми полисомами (и, следовательно, с повышенной скоростью трансляции) по сравнению с аллелем C, содержащим мРНК.
База данных NCBI SNP была проанализирована на предмет наличия вариантов, охватывающих инициирующие кодоны AUG, с акцентом на положения -3 и +4 (Xu et al., 2010). Это исследование выявило SNP в> 45 генах, которые встречаются в одном из этих двух критических положений и, таким образом, потенциально могут влиять на использование AUG. Варианты двух генов были протестированы путем трансфекции репортерных конструкций в клетки и выявили, что мРНК, несущие SNP с «более слабой» или «более сильной» консенсусной последовательностью Козака, вызывают пониженные или повышенные уровни белка, соответственно (Xu et al., 2010). Никаких различий в уровнях мРНК не отмечено.
SNP, создание внутрикадрового uAUG
Мутации, которые генерируют стартовые кодоны выше и в рамке с основным инициирующим кодоном открытой рамки считывания, «улавливают» некоторые сканирующие 43S PIC и перенаправляют синтез белка в новый стартовый сайт для производства N-концевых удлиненных белковых продуктов (рис. 1B). см. «In-frame uAUG»).Эффективность, с которой это достигается, частично диктуется контекстом, окружающим новый инициирующий кодон.
Одним из инструментов биоинформатики, который был разработан для категоризации эффектов вариантов на функцию генома, является SnpEff (Cingolani et al., 2012). Этот инструмент аннотирует варианты на основе местоположения генома, включая интронную, нетранслируемую область, сайт сплайсинга, межгенное, несинонимичное кодирование и т. Д. (Cingolani et al., 2012). Среди эффектов, перечисленных SnpEff, — изменения в кодонах инициации (AUG и менее распространенные кодоны CUG и UUG), которые происходят в 5′-лидерной области.Из 297 SNP, которые генерировали новый кодон инициации трансляции в 5′-лидерной области при сравнении двух штаммов Drosophila melanogaster , ~ 25% находились в той же рамке считывания, что и основная ORF (Cingolani et al., 2012), и вырабатывали N -концевально удлиненные полипептиды.
SNP, создание uORF вне кадра с помощью основного ORF
Если мутация генерирует новый стартовый кодон вне рамки с основной ORF AUG, некоторые PIC 43S могут инициироваться в новой восходящей (u) ORF и обходить основную ORF, что приводит к снижению продукции белка из основной продукт, кодируемый ORF (рисунок 1B и таблица 1).Степень изменения маршрута будет частично зависеть от контекста нового инициирующего кодона, а также от проксимальной вторичной структуры AUG (Kozak, 1991; Barbosa et al., 2013).
В качестве примера такого сценария мутация зародышевой линии в гене β-глобина, расположенная на 26 нуклеотидов выше кодона инициатора AUG, приводит к созданию нового, выходящего за рамки рамки uAUG (Oner et al., 1991; Cai et al. др., 1992). Этот uAUG находится в благоприятном контексте последовательности (A в положении -3), и инициация в этих шунтах uAUG рибосомы проходят аутентичный AUG, снижая продукцию β-глобина и приводя к β-талассемии.Необходимо тщательно оценить, влияет ли на стабильность мРНК конкретная 5′-лидерная мутация, а также влияет ли она на фенотип.
Сходный сценарий был задокументирован в гене GTP cyclohydrolase 1 ( GCh2 ), в котором гетерозиготные мутации связаны с Dopa-чувствительной дистонией (DRD) (Armata et al., 2013). Здесь переход зародышевой линии C в T, расположенный на 22 нуклеотида выше сайта начала трансляции, генерирует новый стартовый кодон, который находится вне рамки с нижележащим кодоном GCh2 и AUG, что приводит к снижению продукции GCh2 (Armata et al., 2013). Будет важно расширить эти результаты, чтобы: (i) формально продемонстрировать, что переход C в T приводит к трансляции вновь созданной нКОРС (оценка, которая может быть сделана с помощью отпечатка рибосом) и (ii) продемонстрировать, что переход C в T альтерация приводит к изменению выхода эндогенного белка GCh2.
Влияние, которое этот класс мутаций может оказывать на биологию опухоли, является значительным и иллюстрируется идентификацией мутации зародышевой линии в гене-супрессоре опухоли CDKN2A , картирующем на 34 нуклеотида выше нормального сайта старта (Liu et al., 1999; Орлоу и др., 2007). В этом случае мутация G в T создает новый инициирующий кодон, находящийся в благоприятном контексте, но вне рамки с CDKN2A AUG. Таким образом, аллель T генерирует мРНК, которая продуцирует меньше CDKN2A и существенно увеличивает риск меланомы у носителей (Liu et al., 1999; Orlow et al., 2007).
SNP, создающие uORF перед основной ORF
Если присутствие SNP приводит к созданию нового uORF, это может повлиять на экспрессию гена: (i) влияя на эффективность повторной инициации в основной нижней рамочной рамке считывания и (ii) генерируя новый пептид, кодируемый uORF (Barbosa и другие., 2013). Точный механизм того, как 40S-рибосомы способны возобновить синтез белка после трансляции нКРС, не совсем хорошо определен, но он связан с длиной нКРС (чем длиннее нКОРС, тем менее эффективен процесс повторной инициации), а также с наличие структурных барьеров в uORF (что снижает потенциал повторной инициации) (рис. 1B; Kozak, 1987b; Abastado et al., 1991; Poyry et al., 2004). Считается, что факторы инициации, критические для повторной инициации, остаются связанными с рибосомами в течение некоторого времени после начала элонгации, но в какой-то момент теряются или выбрасываются из транслирующей рибосомы.Если прекращение трансляции происходит до того, как эти факторы потеряны, то эта рибосома сохраняет свою способность к повторной инициации (Poyry et al., 2004). Анализ 11649 согласованных измерений мРНК и белков из четырех опубликованных исследований на млекопитающих показал, что присутствие uORFs в 5′-лидерной области обычно связано со сниженной экспрессией из основной ORF (Calvo et al., 2009).
Репрессивная природа вновь созданной uORF может частично происходить из-за пониженной эффективности, связанной с повторной инициацией трансляции по сравнению с de novo , cap-зависимой инициацией трансляции.Calvo et al. (2009) предприняли поиск вариантов нуклеотидов, изменяющих ORF, в пределах 12 миллионов SNP, присутствующих в dbSNP. Они обнаружили ряд новых и ранее описанных полиморфизмов, которые, по прогнозам, создают новые или удаляют существующие нКРС. Например, мутации в 5′-лидерной области мРНК SRY (Poulat et al., 1998) и SPINK1 (Witt et al., 2000) вводили новые uORF перед основной ORF. Тестирование репортеров с разными 5′-лидерами показало, что те, которые содержат uORFs, продуцируют меньше основного белка ORF по сравнению с репортерами, экспрессирующими контрольные 5′-лидерные последовательности дикого типа.В общем, возникновение новой uORF связано с 30-80% снижением синтеза белка из основной ORF (Calvo et al., 2009).
Мутации, которые приводят к потере uORF, могут увеличить результат трансляции. Анализ 404 uORF, присутствующих в 5′-лидерах мРНК, кодирующих 83 тирозинкиназы и 49 других протоонкогенов в 308 злокачественных новообразованиях человека, выявил мутации uORF в генах EPHB1 и MAP2K6 (Schulz et al., 2018). В случае EPHB1 мутация изменила единственный uAUG, обнаруженный в 5′-лидере, на кодон GUG, в то время как единственный uAUG из MAP2K6 был изменен на кодон ACG.Обе идентифицированные мутации приводят к увеличению трансляции соответствующих мРНК. Это было дополнено компьютерным анализом данных секвенирования всего экзома из 464 раковых опухолей толстой кишки, который выявил соматические мутации, приводящие к потере 22 кодонов инициации uORF и 31 кодонов терминации uORF (Schulz et al., 2018).
Было также показано, что присутствие uORF придает устойчивость к воздействию цисплатина за счет облегчения трансляции полипептида, кодируемого основной ORF, в стрессовых условиях (Somers et al., 2015; Рисунок 1С). Ген ERCC5 кодирует эндонуклеазу, которая расщепляет 3′-аддукты ДНК и требуется для эксцизионной репарации нуклеотидов. 5′-лидерная область мРНК содержит uORF. Существует полиморфизм G / A, rs751402, расположенный выше этого uORF, где мРНК, содержащая аллель A, имеет новую uORF, а мРНК, содержащую аллель G, нет. Обработка клеток цисплатином приводит к индукции стрессовой реакции, что приводит к фосфорилированию eIF2α и более длительной персистенции трансляции мРНК аллеля А (Somers et al., 2015). В то время как фосфорилирование eIF2α обычно связано с глобальным отключением синтеза белка, трансляционный выход некоторых мРНК парадоксальным образом увеличивается из-за конфигурации uORF в их 5′-лидерных областях.
eIF2 необходим для образования тройного комплекса (с тРНК и GTP). Когда субъединица eIF2α становится фосфорилированной, образование тройного комплекса становится ограничивающим скорость, что приводит к глобальному отключению общей трансляции. Повторная инициация рибосомы после трансляции uORF должна привлекать de novo тройных комплексов и увеличение расстояния uORF до следующего нижестоящего кодона AUG дает больше времени для того, чтобы это событие произошло (Figure 1C).В случае мРНК, содержащей аллель ERCC5 A, создание uORF приводит к тому, что в условиях стресса большинство рибосом, завершивших трансляцию A-кодируемой uORF, не будут повторно приобретать другой тройной комплекс до того, как просканировали прошлое. uORF2 (и, следовательно, uORF2 не будет транслироваться), но сделает это до достижения ORF ERCC5 (рисунок 1C). Создание новых uORFs и их расположение в 5′-лидерной области может, таким образом, изменять то, как трансляция специфических мРНК реагирует на сигнальные сигналы.
SNP, влияющие на область кодирования uORF
Мутации, возникающие в кодирующей области uORFs, могут оказывать два типа эффектов на трансляцию — влияя на природу кодируемого регуляторного пептида и изменяя скорость элонгации.
Если они нарушают функцию регуляторного пептида, участвующего в определении скорости повторной инициации рибосом, то они могут влиять на выход основной ORF. Так может быть G / A SNP в 5′-лидере мРНК трансформирующего фактора роста β3 ( TGFβ3 ) и присутствует у нескольких представителей обширной родословной с аритмогенной кардиомиопатией правого желудочка типа 1 (Beffagna et al., 2005). 5′-лидер мРНК TGFβ3 содержит 11 AUG, потенциально кодирующих 11 полипептидов (Arrick et al., 1991). G / A SNP не изменяет конфигурацию uORF, а скорее вызывает замену Arg на His в кодоне 36 uORF из 88 аминокислот, который находится вне рамки считывания и перекрывается с последовательностью основного AUG TGFβ3 . При тестировании в контексте анализа репортерной люциферазы в трансфицированных клетках миобластов C2C12 присутствие варианта A приводило к 2,5-кратному увеличению продукции люциферазы (Beffagna et al., 2005). Сходная ситуация была описана для гена рецептора серотонина, HTR3A , где C / T SNP, расположенный во втором uORF, вызывал изменение Pro на Ser у людей с биполярным аффективным расстройством (Niesler et al., 2001). Авторы проверили последствия этого SNP в анализе трансфекции на основе люциферазы и обнаружили, что аллель T вызывает 2,5-2,9-кратное увеличение экспрессии люциферазы без изменения уровней мРНК. Одна интерпретация этих результатов состоит в том, что кодируемый uORF пептид играет ингибирующую роль в трансляции, а изменение G на A снижает активность этого небольшого полипептида.В обоих вышеупомянутых исследованиях необходимо оценить потенциальные эффекты SNP на сплайсинг, чтобы исключить другие возможные объяснения наблюдаемых эффектов.
Альтернативно, если варианты влияют на скорость элонгации uORF, то они могут влиять на возможность повторной инициации нижележащими кодонами AUG (Jackson et al., 2012; Gunisova et al., 2018). Считается, что замедление скорости удлинения рибосом, проходящих транзитом через uORF, увеличивает вероятность того, что факторы инициации, связанные с удлинением рибосом и необходимые для удлинения, будут высвобождены до завершения трансляции uORF.Затем это привело бы к снижению повторной инициации нижележащими кодонами AUG. Напротив, если скорости удлинения в uORF увеличиваются, это может привести к увеличению скорости повторной инициации и продукции белка из нижележащих ORF.
5 ‘SNP и активность IRES
Другой способ, с помощью которого изменения последовательности в 5′-лидерных областях, как было документально подтверждено, изменяют трансляцию, — это влияние на активность IRES. Сообщалось, что протоонкоген c-Myc ( MYC ) содержит IRES, который может вносить вклад в неправильную регуляцию трансляции MYC во время онкогенеза (Stoneley et al., 1998). Интересно, что точечная мутация в 5′-лидерной области MYC , приводящая к переходу от C к T, была идентифицирована в линии клеток множественной миеломы и ассоциирована с повышенными уровнями белка MYC (Paulin et al., 1998). 5′-лидер, несущий аллель Т, показал усиленное связывание нескольких связывающих РНК белков, что было выявлено подходами Северо-Западного блоттинга и УФ-перекрестного связывания. Такая же мутация C в T была обнаружена в 42% образцов первичной множественной миеломы и привела к созданию варианта IRES, который оказался более активным (Chappell et al., 2000). Механизм, лежащий в основе того, как изменение C / T может привести к изменениям в деятельности IRES, требует дальнейшего определения.
5′-SNP, выбор сайта инициации транскрипции и альтернативный сплайсинг
Однонуклеотидные полиморфизмы, присутствующие в регуляторных областях генов, могут влиять на фактор транскрипции, а также на связывание РНК Pol II (Kasowski et al., 2010). Если связывание РНК Pol II перенаправляется на вновь образованный сайт, это может привести к использованию альтернативных сайтов инициации транскрипции, генерируя мРНК с различными 5′-лидерными последовательностями, что может повлиять на скорость инициации трансляции.
Вариация последовательности в 5′-лидерной области может также происходить за счет альтернативного сплайсинга мРНК для получения изоформ с различной эффективностью трансляции. Присутствие SNP, влияющих на альтернативный сплайсинг, может изменять уровни и природу образующихся изоформ мРНК. Например, тромбопоэтин ( TPO ) является главным регулятором мегакариопоэза и продукции тромбоцитов и находится под жестким трансляционным контролем. Его 5′-лидер имеет семь uORFs (Ghilardi et al., 1998). Был идентифицирован SNP, который увеличивает сывороточные уровни TPO у пациентов с наследственной тромбоцитемией (Wiestner et al., 1998), генетическое заболевание, вызванное повышенным уровнем тромбоцитов из-за устойчивой пролиферации мегакариоцитов (Murphy et al., 1997). В частности, трансверсия G → C в донорном сайте сплайсинга интрона 3 TPO отвечает за образование укороченного 5′-лидера, в котором uORF 7, а также основной AUG, теряются (Wiestner et al., 1998). Таким образом, инициация трансляции происходит на следующем нижестоящем AUG и приводит к полностью функциональному, хотя и усеченному, белку TPO , уровень которого намного выше, чем у нормальной мРНК.Этот эффект, по-видимому, является результатом повышенной трансляции, предположительно за счет воздействия на процесс повторной инициации, поскольку SNP не влияет на уровни РНК (Wiestner et al., 1998). Влияют ли SNP на сплайсинг или транскрипцию, можно оценить только с помощью характеристики последовательности 5′-лидерных областей мРНК, анализ, который слишком часто опускается.
5′-SNP и сайты-мишени для связывания РНК-белка?
Воздействие на сайты-мишени связывающих РНК белков — еще один способ, с помощью которого SNP могут влиять на трансляцию.Путем измерения соотношения полисомных и связанных с моносомами мРНК в иммортализованных лимфобластоидных клеточных линиях был предпринят полногеномный поиск SNP, влияющих на эффективность трансляции (Li et al., 2013). Это исследование показало, что SNP в 5′-лидерной области мРНК малого рибосомного белка S26 (rs1131017: C / G, расположенный на 22 нуклеотида выше кодона инициатора AUG) был связан с измененной продукцией белка. Репортерные мРНК, содержащие вариант G, продуцировали больше белка, чем мРНК, содержащие вариант C.Этот SNP находится в неравновесном сцеплении с локусом 12q13 для восприимчивости к диабету I типа. Он прерывает полипиримидиновый трек (…. -28 TCTCCT [C / G] TCTCC -17 …) перед кодоном rpS26 AUG. Меняет ли это связывание РНК-связывающего белка, такого как белок, связывающий полипиримидиновый тракт (который участвует в инициации трансляции), еще предстоит определить (Kaminski and Jackson, 1998).
SNP и коэффициенты удлинения
Информация, содержащаяся в мРНК, кодирующей протеом, зашифрована 61 возможным кодоном.Кодоны, кодирующие одну и ту же аминокислоту, декодируются родственными тРНК, которые не одинаково экспрессируются в клетках. Обычно считается, что скорость декодирования кодонов может варьироваться в зависимости от количества тРНК, и это может иметь драматические эффекты на скорость удлинения (Cannarozzi et al., 2010; Hanson and Coller, 2018). Это было подтверждено данными отпечатков рибосом и экспериментами, в которых выход трансляции был увеличен простой заменой редких кодонов на более частые (Gardin et al., 2014; Lareau et al., 2014; Hussmann et al., 2015; Weinberg et al., 2016). Однако считается, что редкие кодоны играют важную роль в клеточном гомеостазе, поскольку участки редких кодонов вызывают паузу рибосом во время удлинения, и это дает время для правильного сворачивания белка (Hanson and Coller, 2018). Таким образом, SNP, изменяющий редкий кодон на более распространенный, может, в принципе, увеличить выход белка, но снизить долю синтезируемого функционального (т.е. правильно свернутого продукта) полипептида.
Примером, когда использование кодонов может влиять на активность белка, можно привести исследование, оценивающее влияние синонимичного SNP (C3435T), присутствующего в кодирующей области множественной лекарственной устойчивости 1 ( MDR1 ), на функцию белка (Kimchi-Sarfaty et al., 2007). Ген MDR1 кодирует управляемый АТФ насос оттока лекарств, который способствует устойчивости опухолевых клеток к лекарствам. SNP C3435T ранее был связан со снижением экспрессии и функции MDR1 в клетках человека (Hoffmeyer et al., 2000; Drescher et al., 2002). Этот SNP заменяет наиболее распространенный кодон Ile (AUC) на менее распространенный (AUU). Репортерные конструкции, содержащие варианты C или T, демонстрируют сходные уровни экспрессии белка, но продуцируют продукты с различной активностью (Kimchi-Sarfaty et al., 2007). Эксперименты по расщеплению трипсином показали, что продукт MDR1 из двух разных гаплотипов различается по чувствительности к протеазам, что указывает на различные конформации. Превращение Ile-кодона во все более редкий, AUA, генерировало мРНК, которая продуцировала белок MDR1 с еще меньшей активностью транспорта лекарств.
Аналогичное явление наблюдалось для гена трансмембранного регулятора проводимости муковисцидоза ( CFTR ), в котором синонимичный SNP T2562G в кодирующей области, как было обнаружено, снижает уровни белка на 30%, не влияя на уровни мРНК или сплайсинг (Kirchner et al., 2017). Этот SNP изменил кодон треонина с широко распространенной последовательности ACU на более редкий кодон ACG. Вектор экспрессии CFTR , несущий аллель G, показал пониженную одноканальную функцию проводимости Cl — по сравнению с вектором, экспрессирующим аллель T (Kirchner et al., 2017). Авторы пришли к выводу, что более низкая скорость синтеза мРНК, кодируемой аллелем G, приводит к неправильному сворачиванию белка, который нацелен на CFTR для деградации механизмом контроля качества (Kirchner et al., 2017). Сниженные уровни белка из мРНК аллеля G были спасены трансфекцией вектора экспрессии, управляющего синтезом SNP-соответствующей родственной тРНК (Kirchner et al., 2017).
Вариация последовательности в 3 ‘Utrs, влияющая на трансляцию
За исключением мРНК гистонов, клеточные мРНК имеют поли (A) хвосты на их 3′-конце. Поли (А) хвост важен для инициации трансляции, и его функция обеспечивается связывающим поли (А) белком, PABPC1. PABPC1 также взаимодействует с eIF4G на 5′-конце мРНК для создания замкнутой петли мРНК, которая, как считается, стимулирует трансляцию посредством: (i) стабилизации ассоциации eIF4F с кэпом, (ii) стимуляции связывания 60S рибосомной субъединицы и ( iii) увеличение эффективной концентрации терминирующих рибосом вблизи кэп-структуры.SNP, которые мутируют сигнал полиаденилирования, будут приводить к генерации изоформ с более длинными 3′-концами из-за использования нижележащих сайтов полиаденилирования (Thomas and Saetrom, 2012). Если расширенная последовательность приводит к приобретению новых сайтов связывания микроРНК (miRNAs), тогда регуляция новой изоформы мРНК может сильно отличаться от мРНК дикого типа (Sandberg et al., 2008).
Также мутации, которые происходят в сайтах-мишенях miRNA и изменяют распознавание miRNA, могут оказывать влияние на экспрессию мРНК за счет снижения инициации трансляции и увеличения деградации мРНК (Mohr and Mott, 2015).За последнее десятилетие мы увидели обширный список SNP, которые отображаются на miRNA или их предполагаемые сайты связывания внутри мРНК, которые потенциально могут влиять на ответ miRNA, и они были всесторонне рассмотрены (Detassis et al., 2017; Moszynska et al., 2017). Например, SNP G к A был описан в miR-1269, miRNA, связанном с повышенным риском гепатоцеллюлярной карциномы (Min et al., 2017). SPATS2L и LRP6 кодируют проонкогенные активности и оба являются мишенями для miR-1269.Это исследование показало, что когда вариант miR-1269 A экспрессируется в клетках, репрессивный эффект на SPATS2L и LRP6 снижается по сравнению с вариантом miR-1269 G. Также были зарегистрированы SNP в сайтах-мишенях микроРНК на мРНК. Например, было обнаружено, что SNP от A до G в 3 ‘UTR мРНК TOMM20 связан с повышенным риском колоректального рака (Lee et al., 2016). Было установлено, что микроРНК miR-4273-5p отвечает за контроль уровней TOMM20 .
Существует несколько примеров белков, связывающих РНК 3′-UTR, которые могут влиять на трансляцию мРНК; как на стадии инициации, так и на стадии удлинения (Szostak, Gebauer, 2013; Yamashita, Takeuchi, 2017). Лучшим примером является 4EHP (также известный как eIF4E2), белок, связывающий кэп, который, как известно, также взаимодействует со специфическими мРНК-связанными белками, присутствующими в 3′-UTR мРНК. Таким образом, 4E-HP образует структуру с замкнутой петлей, и, поскольку он не взаимодействует с eIF4G, это предотвращает рекрутирование рибосом в структуру кэпа и оказывает избирательное ингибирование трансляции мРНК (Morita et al., 2012; Шостак и Гебауэр, 2013; Chapat et al., 2017; Ямасита и Такеучи, 2017). SNP, влияющие на РНК-связывающие белки, которые взаимодействуют с 4EHP, могут приводить к изменениям экспрессии специфического набора 4EHP-чувствительных мРНК.
Заключение
Несмотря на то, что значительные усилия были приложены к поиску и аннотированию SNP, которые могут влиять на функцию белка с использованием таких программ, как SIFT (Kumar et al., 2009) и PolyPhen (Adzhubei et al., 2010; Li and Wei, 2015), существует признанная потребность в инструменте биоинформатики, который может прогнозировать потенциальные функциональные последствия SNP в 5′-лидерных и 3′-UTR мРНК (Kumar et al., 2014). Были достигнуты успехи (i) в отношении программного обеспечения, которое предсказывает эффекты SNP на мишени miRNA, с такими программами, как microSNiPer (Barenboim et al., 2010) и mrSNP (Deveci et al., 2014), (ii) идентификация инициации трансляции. сайты, использующие ATGpr, и (iii) программное обеспечение для прогнозирования ORF, такое как ORF Finder. Теперь необходимы такие инструменты, как SnpEff, которые могли бы связывать изменения в 5′-лидерных и 3′-UTR последовательностях с предсказаниями основной экспрессии ORF. Лучшее понимание переменных, участвующих в определении эффективности трансляции мРНК, поможет разработать алгоритмы с большей количественной предсказательной силой.
Многое было извлечено из функционального анализа генетических вариантов в 5′-лидерах мРНК и их влияния на трансляцию. Большинство из них было идентифицировано, потому что они были связаны с наблюдаемым фенотипом. Поражения, эффекты которых легче всего предсказать, — это те, которые влияют на контекст инициирующих кодонов или приводят к генерации новых uAUG. Однако именно те эффекты, эффекты которых остаются необъясненными, вероятно, приведут к открытию новых биологических механизмов.Например, во время поиска онкогенных изменений, связанных с раком простаты, Wang et al. (2009) идентифицировали соматическую мутацию G в A, которая картировалась в лидерной области 5′-катенина, на девять нуклеотидов выше кодона AUG. Присутствие аллеля А в репортерных мРНК приводило к трех-семикратному увеличению экспрессии белка по сравнению с мРНК, несущими аллель G, без отмеченного влияния на уровни мРНК. Механизм, лежащий в основе этой трансляционной стимуляции, неизвестен, но указывает на очень интересную биологию.Это также подчеркивает необходимость тщательного рассмотрения функциональных последствий 5′-лидерных мутаций, обнаруженных крупномасштабными проектами секвенирования генома рака, и их потенциальной роли в влиянии трансляции. В настоящее время это сложно делать систематически из-за недостатка нашей способности предсказывать структурную сложность РНК, а также из-за недостатка знаний о ландшафте связывающих РНК белков (RBP) in vivo . Чтобы заполнить этот пробел, предпринимаются общегеномные подходы к зондированию структуры РНК, а также усилия, направленные на определение взаимодействия RBP (Castello et al., 2013; Tenzer et al., 2013; Bevilacqua et al., 2016; Бизоньо и Кин, 2018).
Авторские взносы
Все авторы подготовили и написали рецензию.
Финансирование
Исследования в лаборатории JP поддерживаются Канадскими институтами исследований в области здравоохранения (CIHR FDN # 148366).
Заявление о конфликте интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могут быть истолкованы как потенциальный конфликт интересов.
Сноски
- https://www.encodeproject.org/
- https://www.ncbi.nlm.nih.gov/projects/SNP/
Список литературы
Абастадо, Дж. П., Миллер, П. Ф., Джексон, Б. М., и Хиннебуш, А. Г. (1991). Подавление повторной инициации рибосом в вышележащих открытых рамках считывания в клетках с недостатком аминокислот формирует основу для контроля трансляции GCN4. Мол. Клетка. Биол. 11, 486–496. DOI: 10.1128 / MCB.11.1.486
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Аджубей, И.А., Шмидт, С., Пешкин, Л., Раменский, В. Е., Герасимова, А., Борк, П. и др. (2010). Метод и сервер для предсказания повреждающих миссенс-мутаций. Нат. Методы 7, 248–249. DOI: 10.1038 / nmeth0410-248
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Armata, I.A., Balaj, L., Kuster, J.K., Zhang, X., Tsai, S., Armatas, A.A., et al. (2013). Допа-чувствительная дистония: функциональный анализ однонуклеотидных замен в 5′-нетранслируемой области GCh2. PLoS One 8: e76975. DOI: 10.1371 / journal.pone.0076975
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Аррик Б. А., Ли А. Л., Гренделл Р. Л. и Деринк Р. (1991). Ингибирование трансляции мРНК трансформирующего фактора роста бета 3 ее 5′-нетранслируемой областью. Мол. Клетка. Биол. 11, 4306–4313. DOI: 10.1128 / MCB.11.9.4306
CrossRef Полный текст | Google Scholar
Барбоса, К., Пейшейро, И., и Ромао, Л. (2013).Регуляция экспрессии генов с помощью открытых рамок считывания и болезней человека. PLoS Genet. 9: e1003529. DOI: 10.1371 / journal.pgen.1003529
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Баренбойм, М., Золтик, Б. Дж., Го, Ю., и Вайнбергер, Д. Р. (2010). MicroSNiPer: веб-инструмент для прогнозирования эффектов SNP на предполагаемые мишени микроРНК. Гум. Мутат. 31, 1223–1232. DOI: 10.1002 / humu.21349
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Беффанья, Г., Occhi, G., Nava, A., Vitiello, L., Ditadi, A., Basso, C., et al. (2005). Регуляторные мутации в гене трансформирующего фактора роста-бета3 вызывают аритмогенную кардиомиопатию правого желудочка типа 1. Cardiovasc. Res. 65, 366–373. DOI: 10.1016 / j.cardiores.2004.10.005
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Бевилаква, П. К., Ричи, Л. Е., Су, З., и Ассманн, С. М. (2016). Полногеномный анализ вторичной структуры РНК. Annu. Преподобный Жене. 50, 235–266. DOI: 10.1146 / annurev-genet-120215-035034
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Cai, S.P., Eng, B., Francombe, W.H., Olivieri, N.F., Kendall, A.G., Waye, J.S., et al. (1992). Две новые мутации бета-талассемии в 5′- и 3′-некодирующих областях гена бета-глобина. Кровь 79, 1342–1346.
Google Scholar
Кальво, С. Э., Пальярини, Д. Дж., И Мутха, В. К. (2009). Открытые рамки считывания, расположенные выше по течению, вызывают повсеместное снижение экспрессии белка и являются полиморфными среди людей. Proc. Natl. Акад. Sci. США 106, 7507–7512. DOI: 10.1073 / pnas.08106
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Каннароцци, Г., Шраудольф, Н. Н., Фати, М., Фон Рор, П., Фриберг, М. Т., Рот, А. С. и др. (2010). Роль порядка кодонов в динамике трансляции. Cell 141, 355–367. DOI: 10.1016 / j.cell.2010.02.036
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Кастелло, А., Хорос, Р., Штрейн, К., Fischer, B., Eichelbaum, K., Steinmetz, L.M., et al. (2013). Общесистемная идентификация РНК-связывающих белков путем захвата интерактома. Нат. Protoc. 8, 491–500. DOI: 10.1038 / nprot.2013.020
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Чапат, К., Джафарнеджад, С. М., Матта-Камачо, Э., Хескет, Г. Г., Гелбарт, И. А., Аттиг, Дж. И др. (2017). Cap-связывающий белок 4EHP влияет на подавление трансляции микроРНК. Proc. Natl. Акад. Sci. НАС.А. 114, 5425–5430. DOI: 10.1073 / pnas.1701488114
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Чаппелл, С. А., Лекен, Дж. П., Паулин, Ф. Э., Дескулмейстер, М. Л., Стоунли, М., Саутар, Р. Л. и др. (2000). Мутация в c-myc-IRES приводит к усилению входа во внутренние рибосомы при множественной миеломе: новый механизм дерегуляции онкогена. Онкоген 19, 4437–4440. DOI: 10.1038 / sj.onc.1203791
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Чинголани, П., Platts, A., Wang le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). Программа для аннотирования и прогнозирования эффектов однонуклеотидных полиморфизмов, SnpEff: SNP в геноме штамма Drosophila melanogaster w1118; изо-2; iso-3. Fly 6, 80–92. DOI: 10.4161 / fly.19695
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Детассис С., Грассо М., Дель Весково В. и Денти М. А. (2017). МикроРНК делают вызов персонализированной медицине рака. Фронт. Клетка. Dev. Биол. 5:86. DOI: 10.3389 / fcell.2017.00086
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Дьяченко Л., Слэйд Г. Д., Накли А. Г., Бхаланг К., Сигурдссон А., Белфер И. и др. (2005). Генетическая основа индивидуальных вариаций восприятия боли и развития хронического болевого состояния. Гум. Мол. Genet. 14, 135–143. DOI: 10.1093 / hmg / ddi013
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Дрешер, С., Schaeffeler, E., Hitzl, M., Hofmann, U., Schwab, M., Brinkmann, U., et al. (2002). Полиморфизм гена MDR1 и расположение субстрата Р-гликопротеина фексофенадина. Br. J. Clin. Pharmacol. 53, 526–534. DOI: 10.1046 / j.1365-2125.2002.01591.x
CrossRef Полный текст | Google Scholar
Гардин, Дж., Йесмин, Р., Юровский, А., Цай, Ю., Скиена, С., Футчер, Б. (2014). Измерение средней скорости декодирования 61 смыслового кодона in vivo. eLife 3: e03735.DOI: 10.7554 / eLife.03735
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Гиларди Н., Вистнер А. и Шкода Р. К. (1998). Производство тромбопоэтина подавляется трансляционным механизмом. Кровь 92, 4023–4030.
PubMed Аннотация | Google Scholar
Гунисова С., Хронова В., Мохаммад М. П., Хиннебуш А. Г. и Валасек Л. С. (2018). Пожалуйста, не перерабатывайте! Реинициация трансляции у микробов и высших эукариот. FEMS Microbiol. Ред. 42, 165–192. DOI: 10.1093 / femsre / fux059
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Холл, Дж. М., Ли, М. К., Ньюман, Б., Морроу, Дж. Э., Андерсон, Л. А., Хьюи, Б. и др. (1990). Связь семейного рака груди с ранним началом с хромосомой 17q21. Science 250, 1684–1689. DOI: 10.1126 / science.2270482
CrossRef Полный текст | Google Scholar
Хиннебуш, А.Г., Иванов, И.П., Зоненберг, Н.(2016). Контроль трансляции с помощью 5′-нетранслируемых участков мРНК эукариот. Наука 352, 1413–1416. DOI: 10.1126 / science.aad9868
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Hoffmeyer, S., Burk, O., Von Richter, O., Arnold, H.P., Brockmoller, J., Johne, A., et al. (2000). Функциональные полиморфизмы гена множественной лекарственной устойчивости человека: множественные вариации последовательности и корреляция одного аллеля с экспрессией и активностью P-гликопротеина in vivo. Proc. Natl. Акад. Sci. США 97, 3473–3478. DOI: 10.1073 / pnas.97.7.3473
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Хусманн, Дж. А., Патчетт, С., Джонсон, А., Сойер, С., и Пресс, У. Х. (2015). Понимание предубеждений в экспериментах по профилированию рибосом выявляет признаки динамики трансляции у дрожжей. PLoS Genet. 11: e1005732. DOI: 10.1371 / journal.pgen.1005732
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Джексон, Р.Дж., Хеллен К. У., Пестова Т. В. (2012). Конечные и посттерминационные события в эукариотическом переводе. Adv. Protein Chem. Struct. Биол. 86, 45–93. DOI: 10.1016 / B978-0-12-386497-0.00002-5
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Камински А. и Джексон Р. Дж. (1998). Требование белка связывания полипиримидинового тракта (PTB) для внутренней инициации трансляции РНК кардиовирусов является скорее условным, чем абсолютным. РНК 4, 626–638.DOI: 10.1017 / S1355838298971898
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Kasowski, M., Grubert, F., Heffelfinger, C., Hariharan, M., Asabere, A., Waszak, S.M, et al. (2010). Вариации связывания факторов транскрипции у людей. Наука 328, 232–235. DOI: 10.1126 / science.1183621
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Кимчи-Сарфати, К., О, Дж. М., Ким, И. В., Сауна, З. Э., Кальканьо, А. М., Амбудкар, С.V., et al. (2007). «Тихий» полиморфизм в гене MDR1 изменяет субстратную специфичность. Наука 315, 525–528. DOI: 10.1126 / science.1135308
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Кирхнер, С., Цай, З., Раушер, Р., Кастелич, Н., Андин, М., Чех, А., и др. (2017). Изменение функции белка из-за молчащего полиморфизма, связанного с изобилием тРНК. PLoS Biol. 15: e2000779. DOI: 10.1371 / journal.pbio.2000779
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Козак, М.(1986). Точечные мутации определяют последовательность, фланкирующую кодон инициатора AUG, который модулирует трансляцию эукариотическими рибосомами. Cell 44, 283–292. DOI: 10.1016 / 0092-8674 (86) -2
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Козак М. (1987a). Анализ 5′-некодирующих последовательностей из 699 матричных РНК позвоночных. Nucleic Acids Res. 15, 8125–8148.
Google Scholar
Козак, М. (1987b). Влияние интерцистронной длины на эффективность повторной инициации эукариотическими рибосомами. Мол. Клетка. Биол. 7, 3438–3445.
PubMed Аннотация | Google Scholar
Козак, М. (1991). Структурные особенности мРНК эукариот, которые модулируют инициацию трансляции. J. Biol. Chem. 266, 19867–19870.
PubMed Аннотация | Google Scholar
Кумар А., Раджендран В., Сетумадхаван Р., Шукла П., Тивари С. и Пурохит Р. (2014). Вычислительный SNP-анализ: современные подходы и перспективы на будущее. Cell Biochem.Биофиз. 68, 233–239. DOI: 10.1007 / s12013-013-9705-6
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Кумар П., Хеникофф С. и Нг П. К. (2009). Прогнозирование влияния кодирования несинонимичных вариантов на функцию белка с использованием алгоритма SIFT. Нат. Protoc. 4, 1073–1081. DOI: 10.1038 / nprot.2009.86
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ландер, Э.С., Линтон, Л.М., Биррен, Б., Нусбаум, К., Зоди, М.C., Baldwin, J., et al. (2001). Начальная последовательность и анализ человеческого генома. Природа 409, 860–921. DOI: 10.1038 / 35057062
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ларо, Л. Ф., Хайт, Д. Х., Хоган, Г. Дж., И Браун, П. О. (2014). Четкие стадии цикла элонгации трансляции, выявленные при секвенировании защищенных рибосомами фрагментов мРНК. eLife 3: e01257. DOI: 10.7554 / eLife.01257
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ли, А.Р., Пак, Дж., Юнг, К. Дж., Джи, С. Х., и Ким-Юн, С. (2016). Генетическая вариация rs7930 в сайте-мишени miR-4273-5p связана с риском колоректального рака. Onco Targets Ther. 9, 6885–6895. DOI: 10.2147 / OTT.S108787
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ли, Л., и Вэй, Д. (2015). Инструменты биоинформатики для обнаружения и функционального анализа однонуклеотидных полиморфизмов. Adv. Exp. Med. Биол. 827, 287–310. DOI: 10.1007 / 978-94-017-9245-5_17
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ли, К., Макри, А., Лу, Ю., Маршан, Л., Грабс, Р., Руссо, М. и др. (2013). Полногеномный поиск экзонных вариантов, влияющих на эффективность трансляции. Нат. Commun. 4: 2260. DOI: 10.1038 / ncomms3260
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Лю Л., Дилворт Д., Гао Л., Монзон Дж., Саммерс А., Лассам Н. и др. (1999). Мутация 5 ‘UTR CDKN2A создает аберрантный инициирующий кодон и предрасполагает к меланоме. Нат. Genet. 21, 128–132. DOI: 10.1038 / 5082
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Маркотриджиано, Дж., Ломакин, И. Б., Соненберг, Н., Пестова, Т. В., Хеллен, К. У. и Берли, С. К. (2001). Консервативный домен HEAT в eIF4G управляет сборкой механизма инициации трансляции. Мол. Cell 7, 193–203. DOI: 10.1016 / S1097-2765 (01) 00167-8
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Маккарролл, С.А., Хэднотт, Т. Н., Перри, Г. Х., Сабети, П. К., Зоди, М. К., Барретт, Дж. К. и др. (2006). Распространенные делеционные полиморфизмы в геноме человека. Нат. Genet. 38, 86–92. DOI: 10.1038 / ng1696
CrossRef Полный текст | Google Scholar
Менделл, Дж. Т., и Дитц, Х. С. (2001). Когда сообщение идет наперекосяк: вызывающие заболевание мутации, влияющие на содержание и производительность мРНК. Cell 107, 411–414. DOI: 10.1016 / S0092-8674 (01) 00583-9
PubMed Аннотация | CrossRef Полный текст | Google Scholar
мин, стр., Li, W., Zeng, D., Ma, Y., Xu, D., Zheng, W., et al. (2017). Вариант с одним нуклеотидом в микроРНК-1269a способствует возникновению и развитию гепатоцеллюлярной карциномы путем нацеливания на онкогены SPATS2L и LRP6. Бык. Рак 104, 311–320. DOI: 10.1016 / j.bulcan.2016.11.021
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Морита М., Лер Л. В., Фабиан М. Р., Сиддики Н., Муллин М., Хендерсон В. К. и др. (2012). Новый репрессорный комплекс трансляции 4EHP-GIGYF2 необходим для развития млекопитающих. Мол. Клетка. Биол. 32, 3585–3593. DOI: 10.1128 / MCB.00455-12
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Мошинска А., Геберт М., Коллон Дж. Ф. и Бартошевски Р. (2017). SNP в сайтах-мишенях микроРНК и их потенциальная роль в заболеваниях человека. Open Biol. 7: 170019. DOI: 10.1098 / rsob.170019
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Мерфи С., Петерсон П., Иланд Х. и Ласло Дж. (1997).Опыт группы по изучению истинной полицитемии в отношении эссенциальной тромбоцитемии: окончательный отчет о диагностических критериях, выживаемости и лейкозном переходе при лечении. Семин. Гематол. 34, 29–39.
PubMed Аннотация | Google Scholar
Накли А.Г., Шабалина С.А., Чивилева И.Э., Саттерфилд К., Корчинский О., Макаров С.С. и др. (2006). Гаплотипы катехол-О-метилтрансферазы человека модулируют экспрессию белка, изменяя вторичную структуру мРНК. Наука 314, 1930–1933. DOI: 10.1126 / science.1131262
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Николсон П., Епископосян Х., Метце С., Замудио Ороско Р., Кляйншмидт Н. и Мюлеманн О. (2010). Нонсенс-опосредованный распад мРНК в клетках человека: понимание механизмов, функции вне контроля качества и двойная жизнь факторов NMD. Ячейка. Мол. Life Sci. 67, 677–700. DOI: 10.1007 / s00018-009-0177-1
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Нислер, Б., Flohr, T., Nothen, M. M., Fischer, C., Rietschel, M., Franzek, E., et al. (2001). Связь между 5 ‘UTR-вариантом C178T гена серотонинового рецептора HTR3A и биполярным аффективным расстройством. Фармакогенетика 11, 471–475. DOI: 10.1097 / 00008571-200108000-00002
CrossRef Полный текст | Google Scholar
Онер Р., Агарвал С., Димовски А. Дж., Ефремов Г. Д., Петков Г. Х., Алтай К. и др. (1991). Мутация G-A в положении + 22 3 ‘к сайту Cap бета-глобинового гена как возможная причина бета-талассемии. Гемоглобин 15, 67–76. DOI: 10.3109 / 036302691085
CrossRef Полный текст | Google Scholar
Орлоу И., Бегг К. Б., Котинола Дж., Рой П., Хаммер А. Дж., Клас Б. А. и др. (2007). Мутации зародышевой линии CDKN2A у лиц со злокачественной меланомой кожи. J. Invest. Дерматол. 127, 1234–1243. DOI: 10.1038 / sj.jid.5700689
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Орр, Н., Чанок, С. (2008). Общие генетические вариации и болезни человека. Adv. Genet. 62, 1–32. DOI: 10.1016 / S0065-2660 (08) 00601-9
CrossRef Полный текст | Google Scholar
Полин Ф. Э., Чаппелл С. А. и Уиллис А. Э. (1998). Одно нуклеотидное изменение во внутреннем сегменте входа в рибосому c-myc приводит к усиленному связыванию группы белковых факторов. Nucleic Acids Res. 26, 3097–3103. DOI: 10.1093 / nar / 26.13.3097
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Пеллетье, Дж., Графф, Дж., Руджеро Д., Зоненберг Н. (2015). Ориентация на комплекс инициации трансляции eIF4F: критическая связь для развития рака. Cancer Res. 75, 250–263. DOI: 10.1158 / 0008-5472.CAN-14-2789
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Пеллетье Дж. И Зоненберг Н. (1985). Инсерционный мутагенез для увеличения вторичной структуры в 5′-некодирующей области эукариотической мРНК снижает эффективность трансляции. Cell 40, 515–526.DOI: 10.1016 / 0092-8674 (85)
CrossRef Полный текст | Google Scholar
Пикеринг Б. М., Уиллис А. Э. (2005). Влияние структурированных 5′-нетранслируемых областей на трансляцию и болезнь. Семин. Клетка. Dev. Биол. 16, 39–47. DOI: 10.1016 / j.semcdb.2004.11.006
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Пула, Ф., Десклозо, М., Таффери, С., Джей, П., Бойзе, Б., и Берта, П. (1998). Мутация в 5′-некодирующей области гена SRY у пациента с измененным полом XY. Гум. Мутат. Дополнение 1, S192 – S194. DOI: 10.1002 / humu.1380110162
CrossRef Полный текст | Google Scholar
Пойри, Т. А., Камински, А., Джексон, Р. Дж. (2004). Что определяет, возобновят ли сканирование рибосомы млекопитающих после трансляции короткой вышележащей открытой рамки считывания? Genes Dev. 18, 62–75.
PubMed Аннотация | Google Scholar
Сандберг Р., Нейлсон Дж. Р., Сарма А., Шарп П. А. и Бердж К. Б. (2008). Пролиферирующие клетки экспрессируют мРНК с укороченными 3′-нетранслируемыми областями и меньшим количеством сайтов-мишеней для микроРНК. Наука 320, 1643–1647. DOI: 10.1126 / science.1155390
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Schulz, J., Mah, N., Neuenschwander, M., Kischka, T., Ratei, R., Schlag, P.M, et al. (2018). Мутации с потерей функции uORF при злокачественных новообразованиях человека. Sci. Отчет 8: 2395. DOI: 10.1038 / s41598-018-19201-8
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Шен, Л. X., Базилион, Дж. П., и Стэнтон, В. П. младший.(1999). Однонуклеотидные полиморфизмы могут вызывать различные структурные складки мРНК. Proc. Natl. Акад. Sci. США 96, 7871–7876. DOI: 10.1073 / pnas.96.14.7871
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Синьори, Э., Баньи, К., Папа, С., Примерано, Б., Ринальди, М., Амальди, Ф. и др. (2001). Соматическая мутация в 5’UTR гена BRCA1 при спорадическом раке молочной железы вызывает снижение эффективности трансляции. Онкоген 20, 4596–4600.DOI: 10.1038 / sj.onc.1204620
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Somers, J., Wilson, L.A., Kilday, J.P., Horvilleur, E., Cannell, I.G., Poyry, T.A., et al. (2015). Общий полиморфизм в 5′-UTR ERCC5 создает расположенную выше ORF, которая придает устойчивость к химиотерапии на основе платины. Genes Dev. 29, 1891–1896. DOI: 10.1101 / gad.261867.115
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Зоненберг, Н.и Хиннебуш А.Г. (2009). Регуляция инициации трансляции у эукариот: механизмы и биологические мишени. Cell 136, 731–745. DOI: 10.1016 / j.cell.2009.01.042
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Сринивасан, С., Клементс, Дж. А., и Батра, Дж. (2016). Однонуклеотидные полиморфизмы в клиниках: фантастика или реальность для рака? Крит. Преподобный Clin. Лаборатория. Sci. 53, 29–39. DOI: 10.3109 / 10408363.2015.1075469
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Стоунли, М., Paulin, F.E., Le Quesne, J.P., Chappell, S.A., and Willis, A.E. (1998). Нетранслируемая область C-Myc 5 ‘содержит внутренний входной сегмент рибосомы. Онкоген 16, 423–428. DOI: 10.1038 / sj.onc.1201763
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Сабо, К. И., и Кинг, М. К. (1995). Унаследованный рак груди и яичников. Гум. Мол. Genet. 4, 1811–1817. DOI: 10.1093 / hmg / 4.suppl_1.1811
CrossRef Полный текст | Google Scholar
Тензер, С., Моро, А., Кухарев, Дж., Фрэнсис, А. С., Видалино, Л., Провензани, А. и др. (2013). Полнопротеомная характеристика РНК-связывающего белка RALY-интерактома с использованием подхода in vivo-biotinylation-pulldown-Quant (iBioPQ). J. Proteome Res. 12, 2869–2884. DOI: 10.1021 / pr400193j
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Томас, Л. Ф., и Саэтром, П. (2012). Полиморфизм одиночных нуклеотидов может создавать альтернативные сигналы полиаденилирования и влиять на экспрессию генов за счет потери регуляции микроРНК. PLoS Comput. Биол. 8: e1002621. DOI: 10.1371 / journal.pcbi.1002621
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Wang, T., Chen, Y.H., Hong, H., Zeng, Y., Zhang, J., Lu, J.P., et al. (2009). Повышенные нуклеотидные полиморфные изменения в 5′-нетранслируемой области гена дельта-катенина (CTNND2) при раке простаты. Онкоген 28, 555–564. DOI: 10.1038 / onc.2008.399
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Вайнберг, Д.Э., Шах, П., Эйххорн, С. В., Хусманн, Дж. А., Плоткин, Дж. Б., и Бартель, Д. П. (2016). Улучшенные измерения рибосомного следа и мРНК дают представление о динамике и регуляции дрожжевой трансляции. Cell Rep. 14, 1787–1799. DOI: 10.1016 / j.celrep.2016.01.043
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Вистнер А., Шлемпер Р. Дж., Ван Дер Маас А. П. и Шкода Р. К. (1998). Активирующая мутация донора сплайсинга в гене тромбопоэтина вызывает наследственную тромбоцитемию. Нат. Genet. 18, 49–52. DOI: 10.1038 / ng0198-49
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Witt, H., Luck, W., Hennies, H.C., Classen, M., Kage, A., Lass, U., et al. (2000). Мутации в гене, кодирующем ингибитор сериновой протеазы, Kazal типа 1, связаны с хроническим панкреатитом. Нат. Genet. 25, 213–216. DOI: 10.1038 / 76088
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Сюй, Х., Ван, П., You, J., Zheng, Y., Fu, Y., Tang, Q., et al. (2010). Скрининг SNP, расположенных по мотивам Козака, и анализ их ассоциации с заболеваниями человека. Biochem. Биофиз. Res. Commun. 392, 89–94. DOI: 10.1016 / j.bbrc.2010.01.002
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ямасита А., Такеучи О. (2017). Трансляционный контроль мРНК с помощью связывающих белков 3′-нетранслируемой области. BMB Rep. 50, 194–200. DOI: 10.5483 / BMBRep.2017.50.4.040
CrossRef Полный текст | Google Scholar
Янагия А., Свиткин Ю. В., Шибата С., Миками С., Иматака Х. и Соненберг Н. (2009). Необходимость связывания РНК фактора инициации трансляции эукариот млекопитающих 4GI (eIF4GI) для эффективного взаимодействия eIF4E с кэпом мРНК. Мол. Клетка. Биол. 29, 1661–1669. DOI: 10.1128 / MCB.01187-08
PubMed Аннотация | CrossRef Полный текст | Google Scholar
MGI-Рекомендации по номенклатуре генов, генетических маркеров, аллелей и мутаций у мышей и крыс
Рекомендации по номенклатуре генов, генетических маркеров, аллелей и мутаций у мышей и крыс
Доработана: сентябрь 2021 г.
Международный комитет по стандартизированной генетической номенклатуре мышей
Председатель: Dr.Синтия Смит
(электронная почта: [email protected])
Комитет по геному и номенклатуре крыс
Председатель: Д-р Стэнли Лауледеркинд
Правила генетической номенклатуры мышей были впервые опубликованы Данном, Грюнебергом и Снеллом (1940), а последующие версии были опубликованы Международным комитетом по стандартизированной генетической номенклатуре мышей (1963, 1973, 1981, 1989, 1996). Самую последнюю публикацию рекомендаций по номенклатуре мышей можно найти у Eppig (2006).Однако следует сообщить пользователям, что эта веб-версия представляет собой текущую политику номенклатуры Международного комитета по стандартизированной генетической номенклатуре мышей и имеет прецедент по сравнению с ранее опубликованными версиями.
Правила генетической номенклатуры крыс были впервые опубликованы Комитетом по номенклатуре крыс в 1992 году, а затем Levan et al. в 1995 году.
В 2003 году Международный комитет по стандартизированной генетической номенклатуре мышей и
Комитет по геному и номенклатуре крыс согласился унифицировать правила и рекомендации
для номенклатуры генов, аллелей и мутаций у мышей и крыс.Руководства по номенклатуре в настоящее время ежегодно пересматриваются и обновляются двумя международными комитетами; текущие руководящие принципы могут быть
можно найти на сайтах MGD и RGD.
Чтобы просмотреть предыдущие версии этого руководства (последний раз редактировалось в декабре 2015 г.), щелкните здесь.
Содержание
1 Принципы номенклатуры
1.1 Основные характеристики
1.2 Определения
1.3 Стабильность номенклатуры
1.4 Синонимы
1.5 Символы генов, белки и обозначения хромосом в публикациях
1.5.1 Символы генов и аллелей
1.5.2 Символы белков
1.5.3 Обозначения хромосом
2 Символы и названия генов и локусов
2.1 Лабораторные коды
2.2 Идентификация новых генов
2.3 Символы и названия генов
2.3.1 Символы генов
2.3.2 Названия генов
2.4 Структурные гены, варианты сплайсинга и промоторы
2.4.1 Альтернативные транскрипты
2.4.2 Сквозные транскрипты
2.4.3 Антисмысловые и противоположные гены
2.4.4 Гены с гомологами у других видов
2.5 Названия и символы фенотипов
2.5.1 Летальные фенотипы
2.6 Семейства генов
2.6.1 Семьи, идентифицированные гибридизацией
2.6.2 Семьи, идентифицированные путем сравнения последовательностей
2.7 EST
2.8 Анонимные сегменты ДНК
2.8 .1 Картированные сегменты ДНК
2.8.2 STS, используемые при физическом картировании
2.9 Локусы генной ловушки
2.10 Локусы количественных признаков, гены устойчивости и гены иммунного ответа
2.10.1 Названия и символы QTL
2.10.2 Определение уникальности в QTL
2.11 Хромосомные области
2.11.1 Теломеры
2.11.2 Центромеры и перицентрический гетерохроматин
2.11.3 Организаторы ядрышка
2.11.4 Гомогенно окрашивающие области
2.12 Гены, расположенные на митохондриях
2.13 Гены домашнего хозяйства РНК, закодированные в ядре
2.14 микроРНК и кластеры микроРНК
2.15 Длинные некодирующие РНК
2.16 Энхансеры, промоторы и регуляторные области
3 Названия и символы для вариантов и мутантных аллелей
3.1 Мутантные фенотипы
3.1.1 Гены, известные только мутантным фенотипам
3.1.2 Фенотипы, вызванные мутациями в структурных генах
3.1.3 Дикий тип Аллели и ревертанты
3.2 Варианты
3.2.1 Биохимические варианты
3.2.2 Варианты сегментов ДНК
3.2.3 Однонуклеотидные полиморфизмы (SNP)
3.2.4 Геномный вариант
3.3 Вариация локусов количественных признаков и генов ответа и устойчивости
3.4 Инсерционные и индуцированные мутации
3.4.1 Мутации структурных генов
3.4.2 Трансгенные инсерционные мутации
3.5 Целевые и захваченные мутации
3.5.1 Нокаут, нокаут, условные и другие Целевые мутации
3.5.2 Мутации, опосредованные эндонуклеазами
3.5.3 Мутации генных ловушек
3.5.4 Энхансерные ловушки
4 Трансгены
4.1 Символы трансгенов
4.2 Сайты межгенных вставок, используемые в качестве сайтов посадки «нейтральных» реципиентных последовательностей
5 Мутации и вставки, индуцированные транспозонами
5.1 Конкатамеры трансгенных мобильных элементов (TE)
5.2 Транспозазные вставки
5.3 Транспонированные вставные аллели
6 Определения
6.1 Ген
6.2 Pseudogen
6.3 Локус
6.4 Маркер
6.5 Аллель
6.6 Аллельный вариант
6.7 Вариант сплайсинга или альтернативный сплайс
6.8 Мутация
6.9 Доминантный и рецессивный
6.10 Генотип
6.11 Фенотип
6.12 Локусы количественных признаков (QTL)
6,13 Гаплотип
6,14 Гомолог
6,15 Ортолог
6,16 Паралог
6,17 Кластер
7 Ссылки
1 Принципы номенклатуры
1.1 Основные характеристики
Ключевым компонентом номенклатуры является имя и символ гена или локуса, которые идентифицируют единицу наследования. Другие особенности, такие как аллели, варианты и мутации, вторичны по отношению к названию гена и становятся
связанные с ним.Точно так же зонды или анализы, используемые для обнаружения гена, не являются первичными характеристиками и обычно не должны использоваться в качестве названий.
Основная цель имени и символа гена или локуса — быть уникальным идентификатором, чтобы информация о гене в публикациях, базах данных и других формах
коммуникация может быть однозначно связана с правильным геном. Это руководство, таким образом, предназначено для того, чтобы помочь научному сообществу в целом использовать генетическую информацию.
Прочие второстепенные функции номенклатуры генов:
- идентифицирует ген как член семьи, что может дать дополнительную информацию о гене
со ссылкой на других членов семьи - идентифицировать ген как ортолог гена у другого млекопитающего (обычно человека)
1.2 Определения
Важно, чтобы пользователь понимал, что называется, и принципы, лежащие в основе этих рекомендаций.
В разделе 6 представлены определения, которые помогут пользователю различать, например, гены, локусы, маркеры и аллели.
1.3 Стабильность номенклатуры
В целом названия генов должны быть стабильными; то есть их не следует менять с течением времени. Однако есть определенные обстоятельства, при которых изменение желательно:
- В случаях, когда ген был известен только как мутантный фенотип и назван в его честь: когда мутировавший ген идентифицирован, имя мутанта становится именем мутантного аллеля идентифицированного гена (см.1.2).
- Когда ген назначается семейству генов (паралогов) и устанавливается номенклатура этого семейства. (см. раздел 2.6.2).
- Если ортологичный ген (ы) был идентифицирован между
мышь, крыса и человек, и для всех трех видов принят общий символ.
1.4 Синонимы
У гена может быть несколько синонимов, то есть имен или символов, которые применялись к гену в разное время. Эти синонимы могут быть связаны с геном в базах данных и публикациях, но установленные
имя и символ гена всегда следует использовать в качестве первичного идентификатора.
1.5 Символы генов, белки и обозначения хромосом в публикациях
1.5.1 Символы генов и аллелей
Символы генов при публикации выделяются курсивом, как и символы аллелей. Раздел 2 ниже определяет правила именования для определения правильных символов генов. Трансгены, не являющиеся частью нативного генома, курсивом не выделяются. Для определения правильного назначения символов гена и аллеля доступна помощь ([email protected]), а символы можно зарезервировать перед публикацией в частном порядке.
Чтобы различать формы мРНК, геномной ДНК и кДНК в рукописи, напишите соответствующий префикс в скобках перед символом гена, например (мРНК) Rbp1 .
1.5.2 Белковые символы
Обозначения белков соответствуют тем же правилам, что и символы генов, со следующими двумя различиями:
- В символах белков используются только прописные буквы.
- Символы белков не выделены курсивом.
1.5.3 Обозначения хромосом
- Используйте заглавную букву «C» при обозначении конкретной хромосомы мыши (например,g., хромосома 15) для аутосомных хромосом. Для половых хромосом допускается использование «Х-хромосомы» или «Y-хромосомы».
- При сокращении слова Хромосома не используйте точку («.») После сокращения (например, Хромосома 15 должна сокращаться как Chr 15, а не Chr.15).
2 символа и названия генов и локусов
Основная функция имени гена — предоставить уникальный идентификатор.
База данных генома мышей (MGD) служит центральным хранилищем названий генов и символов, чтобы избежать использования одного и того же имени для разных генов или использования нескольких
названия одного и того же гена (http: // www.informatics.jax.org). Номенклатурный комитет MGD ([email protected]) предоставляет консультации и помощь в присвоении новых имен и символов. Веб-инструмент для предложения нового символа локуса мыши находится на сайте MGD.
Для крыс эти функции выполняет RGD (http://rgd.mcw.edu) при содействии Международного комитета по геному и номенклатуре крыс (RGNC). Веб-инструмент для предложения нового символа локуса крысы находится на сайте RGD.
2.1 Лабораторные коды
Ключевой особенностью номенклатуры мышей и крыс является регистрационный код лаборатории или код лаборатории, который обычно состоит из трех-четырех букв (первая буква в верхнем регистре, а затем все в нижнем регистре), который идентифицирует
конкретный институт, лаборатория или исследователь, которые производили и могут хранить запасы
Например, маркер ДНК, линия мыши или крысы или создатель новой мутации.Лабораторные коды также используются для обозначения хромосомных аберраций, трансгенов и генно-инженерных мутаций. Поскольку коды лабораторий являются ключом к идентификации исходных источников, они присваиваются не «проектам», а скорее конкретному производителю / создателю или сайту.
Коды лабораторий могут быть присвоены через MGD или непосредственно Институтом исследований лабораторных животных (ILAR) по адресу http://dels-old.nas.edu/ilar_n/ilarhome/register_lc.php.
Примеры:
J The Jackson Laboratory Mit Massachusetts
Технологический институтLeh Hans Lehrach Kyo Kyoto University Ztm Central Animal Laboratory Medical School Hannover
2.2 Идентификация новых генов
Идентификация новых генов в целом происходит двумя способами; идентификация нового белка или последовательности ДНК или идентификация нового фенотипа или признака.
В случае последовательностей следует проявлять осторожность при интерпретации результатов поиска в базе данных для установления новизны (например, чтобы различать новый член семейства генов и аллель или альтернативный транскрипт существующего члена семьи).
Новые мутантные фенотипы или признаки должны быть названы в соответствии с их первичной характеристикой, но как только ген, ответственный за фенотипические вариации, идентифицирован, это дает первичное название гена, а название мутанта становится названием аллеля (см. Раздел 2.3).
2.3 Символы и названия генов
2.3.1 Генные символы
Гены обозначены короткими символами в качестве удобных сокращений для того, чтобы говорить и писать о генах.
A символ гена должен:
- быть уникальным в пределах данного вида и не должно совпадать с символом у другого вида, который
не является гомологом. - быть коротким, обычно 3-5 знаков, но не более 10 знаков
- использовать
только латинские буквы и арабские цифры - начинается с заглавной буквы (не числа), за которой следуют все строчные буквы / цифры (см. Исключение ниже)
- без обозначения тканевой специфичности или молекулярной массы
- включать знаки препинания только в особых случаях (см. Ниже)
- иметь ту же начальную букву, что и начальная буква названия гена, чтобы облегчить индексацию.Однако порядок букв в символе гена не обязательно должен соответствовать порядку слов в имени.
- Когда существует окончательный человеческий ортолог, символы генов должны согласовываться с символами человеческих генов, когда это возможно.
- Никогда не добавляйте «m» (для символа гена мыши) или «r» (для символа гена крысы), чтобы указать вид.
В идеале
Примеры:
Plaur Активатор плазминогена, рецептор урокиназы Sta аутосомное чередование
- в опубликованных статьях выделяться курсивом.Поскольку их трудно читать, в зависимости от браузера, символы генов часто не выделяются курсивом при размещении на веб-странице
- использует общий символ основы или корня, когда принадлежит к семейству генов. Номера членов семьи или обозначения субъединиц следует помещать в конце символа гена.
Примеры: